

Structural and sequencing analysis of local target DNA recognition by MLV integrase
- PMID: 25969444
- PMCID: PMC4477651
- DOI: 10.1093/nar/gkv410
Structural and sequencing analysis of local target DNA recognition by MLV integrase
Abstract
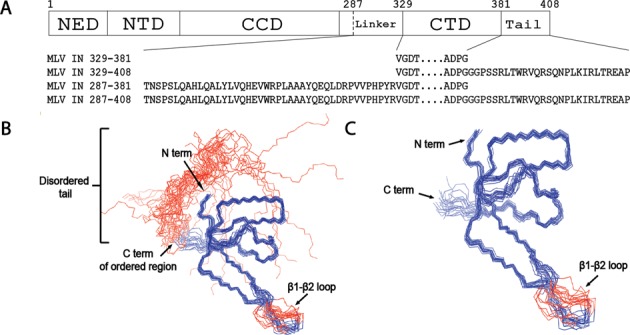
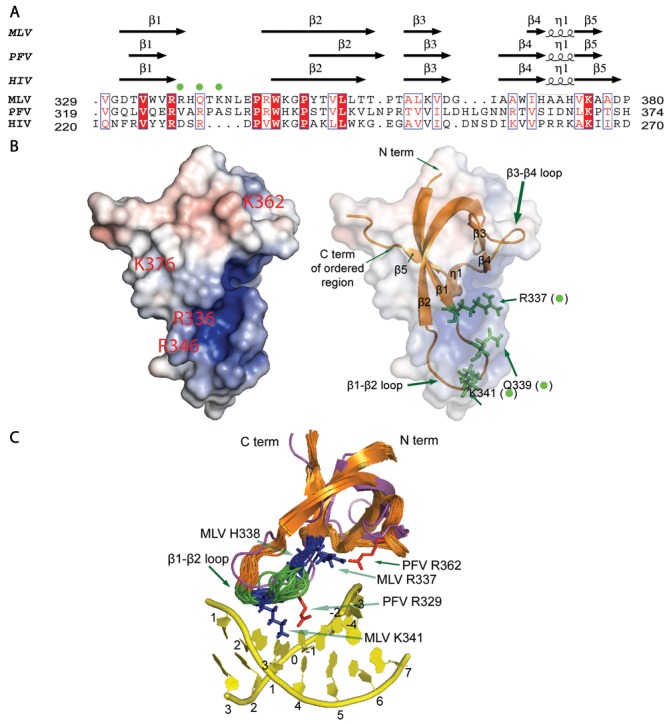
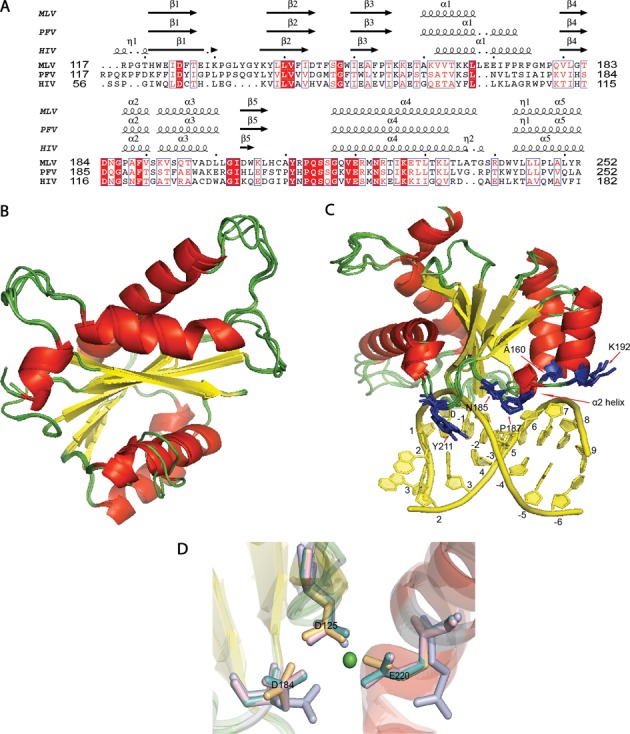
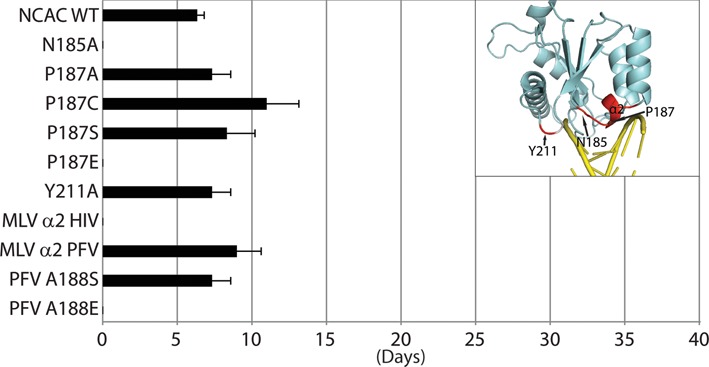
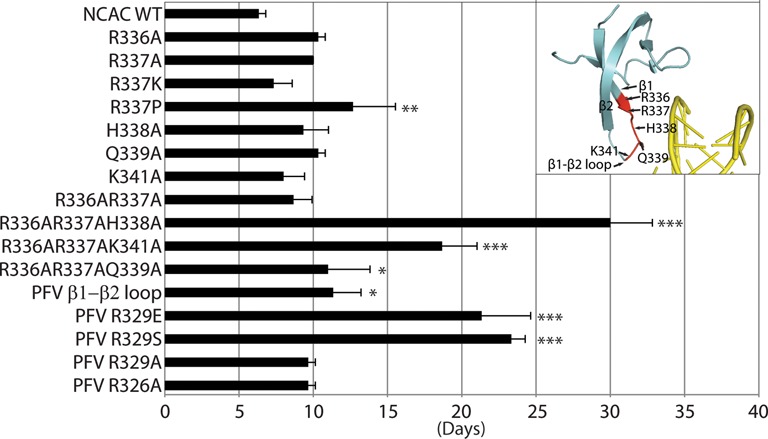
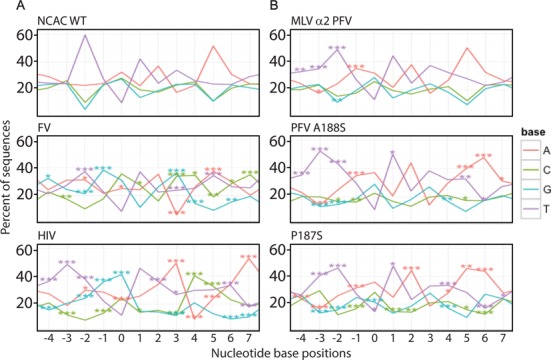
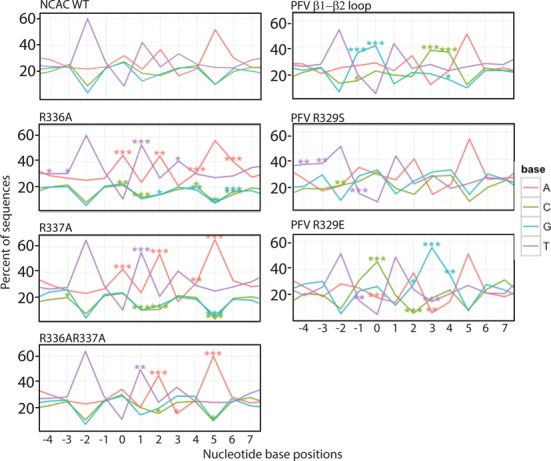
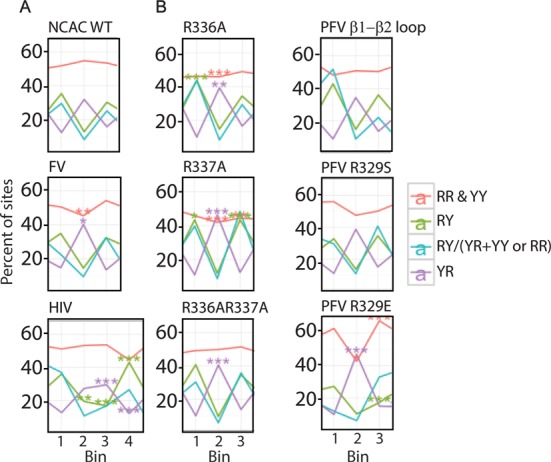
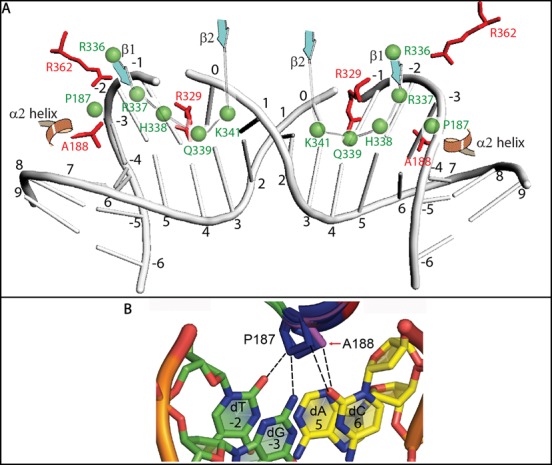
Target-site selection by retroviral integrase (IN) proteins profoundly affects viral pathogenesis. We describe the solution nuclear magnetic resonance structure of the Moloney murine leukemia virus IN (M-MLV) C-terminal domain (CTD) and a structural homology model of the catalytic core domain (CCD). In solution, the isolated MLV IN CTD adopts an SH3 domain fold flanked by a C-terminal unstructured tail. We generated a concordant MLV IN CCD structural model using SWISS-MODEL, MMM-tree and I-TASSER. Using the X-ray crystal structure of the prototype foamy virus IN target capture complex together with our MLV domain structures, residues within the CCD α2 helical region and the CTD β1-β2 loop were predicted to bind target DNA. The role of these residues was analyzed in vivo through point mutants and motif interchanges. Viable viruses with substitutions at the IN CCD α2 helical region and the CTD β1-β2 loop were tested for effects on integration target site selection. Next-generation sequencing and analysis of integration target sequences indicate that the CCD α2 helical region, in particular P187, interacts with the sequences distal to the scissile bonds whereas the CTD β1-β2 loop binds to residues proximal to it. These findings validate our structural model and disclose IN-DNA interactions relevant to target site selection.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Engelman A., Mizuuchi K., Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. - PubMed
-
- Wu X., Li Y., Crise B., Burgess S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. - PubMed
-
- Dyda F., Hickman A.B., Jenkins T.M., Engelman A., Craigie R., Davies D.R. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
