

Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone
- PMID: 25970247
- PMCID: PMC10601608
- DOI: 10.1038/nature14490
Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone
Erratum in
-
Erratum: Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone.Nature. 2015 Oct 22;526(7574):595. doi: 10.1038/nature15255. Epub 2015 Aug 26. Nature. 2015. PMID: 26308898 Free PMC article. No abstract available.
Abstract
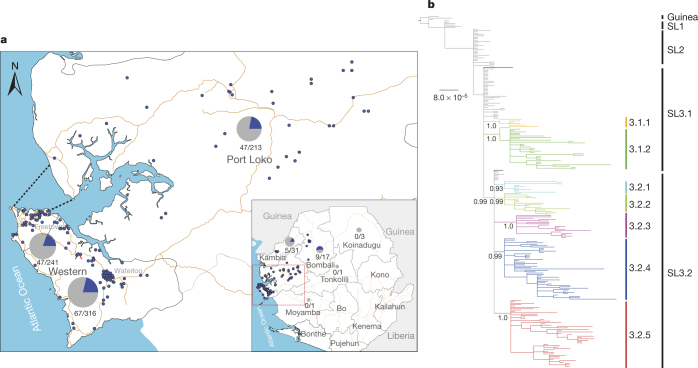
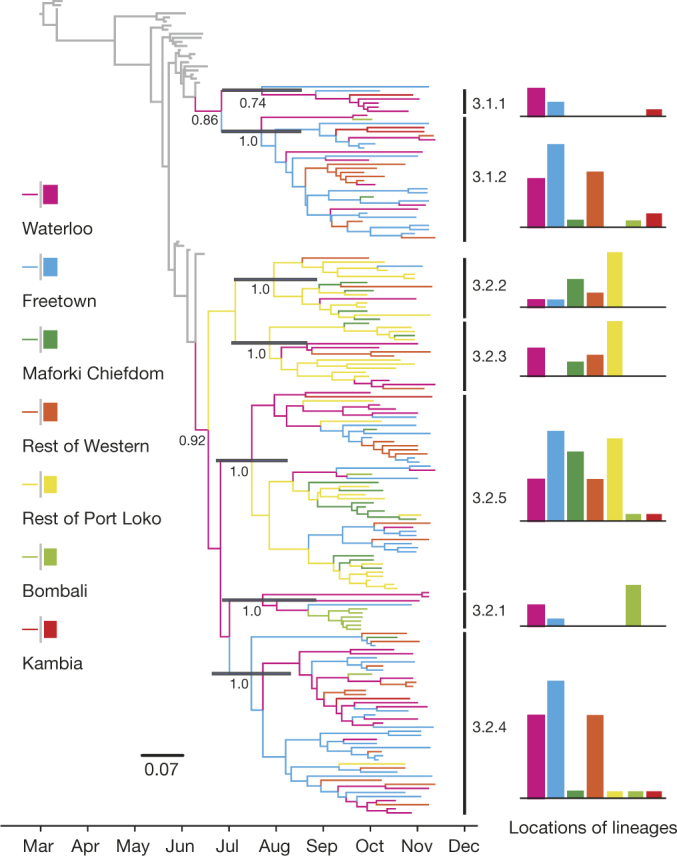
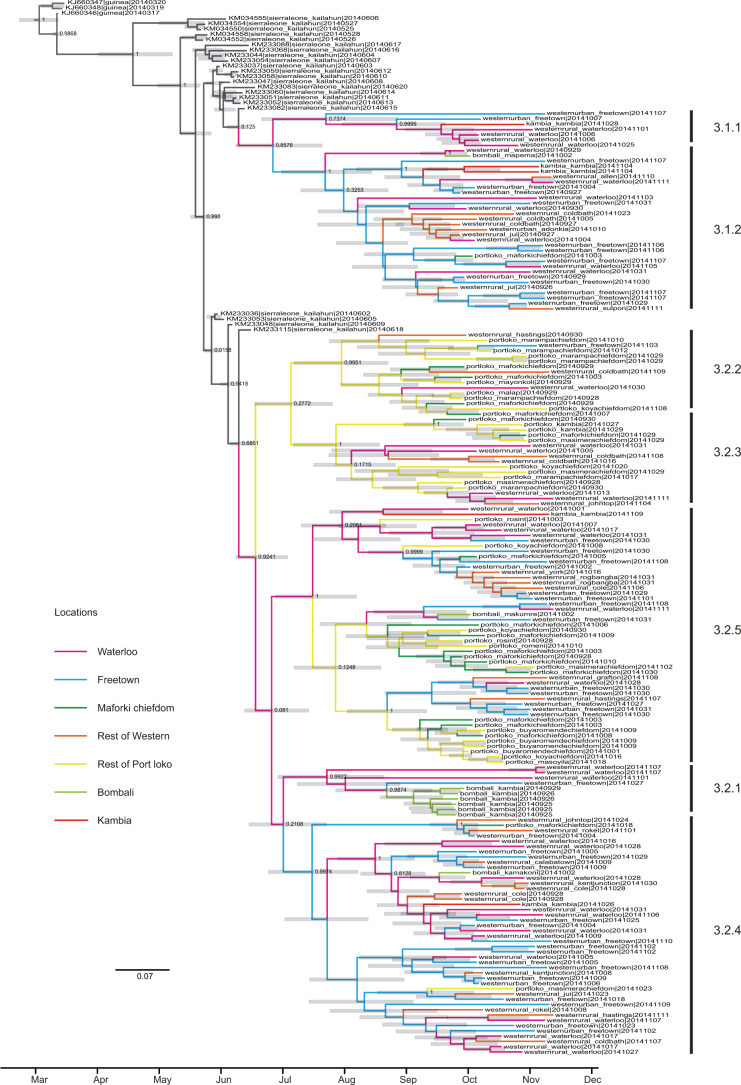
A novel Ebola virus (EBOV) first identified in March 2014 has infected more than 25,000 people in West Africa, resulting in more than 10,000 deaths. Preliminary analyses of genome sequences of 81 EBOV collected from March to June 2014 from Guinea and Sierra Leone suggest that the 2014 EBOV originated from an independent transmission event from its natural reservoir followed by sustained human-to-human infections. It has been reported that the EBOV genome variation might have an effect on the efficacy of sequence-based virus detection and candidate therapeutics. However, only limited viral information has been available since July 2014, when the outbreak entered a rapid growth phase. Here we describe 175 full-length EBOV genome sequences from five severely stricken districts in Sierra Leone from 28 September to 11 November 2014. We found that the 2014 EBOV has become more phylogenetically and genetically diverse from July to November 2014, characterized by the emergence of multiple novel lineages. The substitution rate for the 2014 EBOV was estimated to be 1.23 × 10(-3) substitutions per site per year (95% highest posterior density interval, 1.04 × 10(-3) to 1.41 × 10(-3) substitutions per site per year), approximating to that observed between previous EBOV outbreaks. The sharp increase in genetic diversity of the 2014 EBOV warrants extensive EBOV surveillance in Sierra Leone, Guinea and Liberia to better understand the viral evolution and transmission dynamics of the ongoing outbreak. These data will facilitate the international efforts to develop vaccines and therapeutics.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
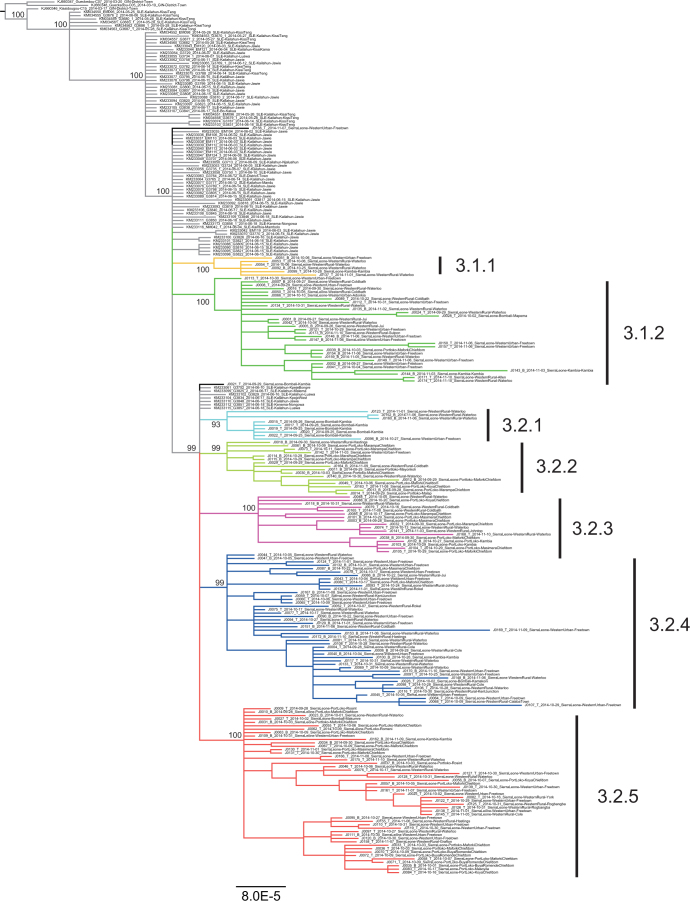
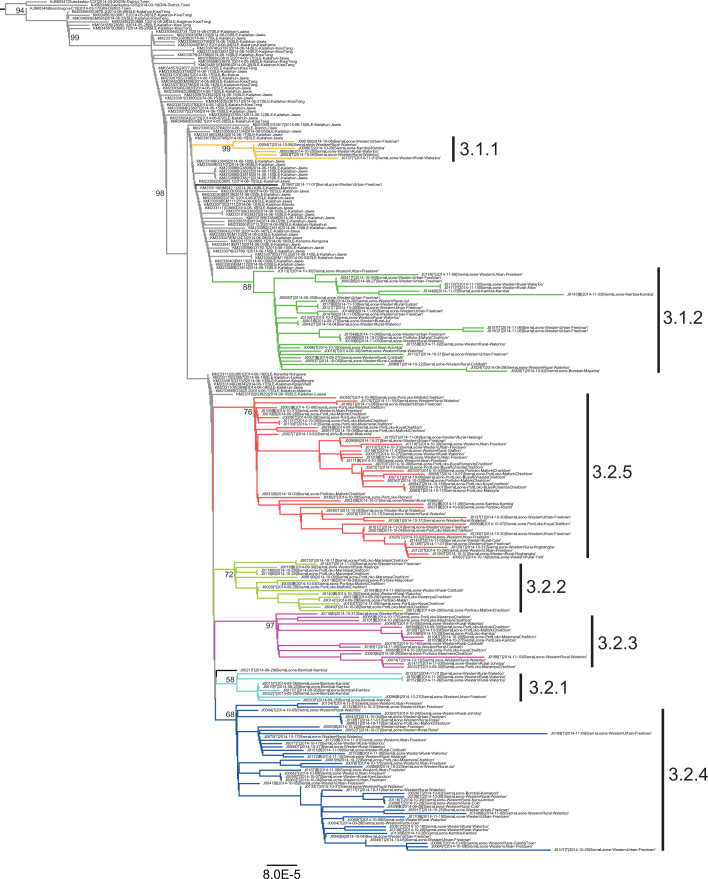
Comment in
-
Inside the Chinese lab poised to study world's most dangerous pathogens.Nature. 2017 Feb 22;542(7642):399-400. doi: 10.1038/nature.2017.21487. Nature. 2017. PMID: 28230144 No abstract available.
References
-
- World Health Organization. Ebola response roadmap - Situation report. http://www.who.int/csr/disease/ebola/situation-reports/en (accessed, 1 April 2015)
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
