

Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology
- PMID: 25970732
- PMCID: PMC4534171
- DOI: 10.1002/pro.2696
Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology
Abstract
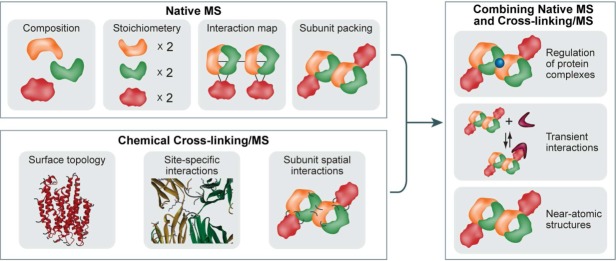
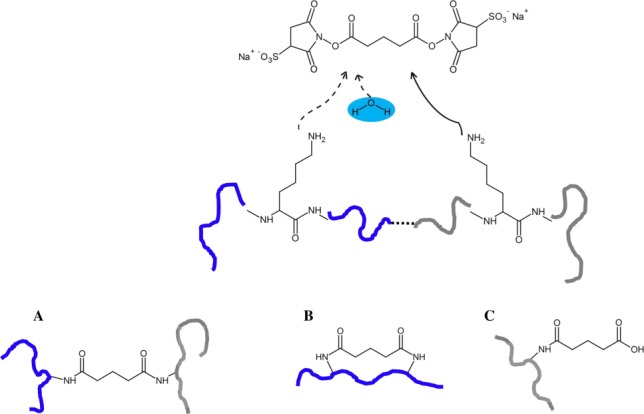
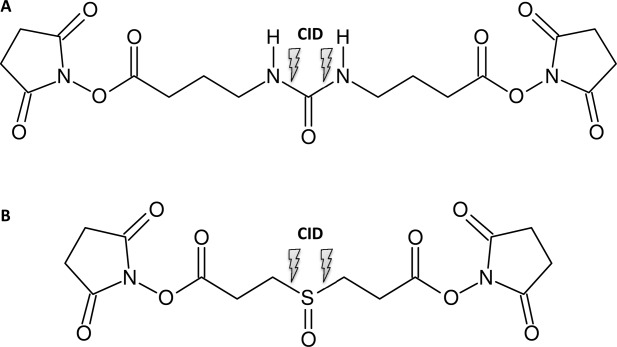
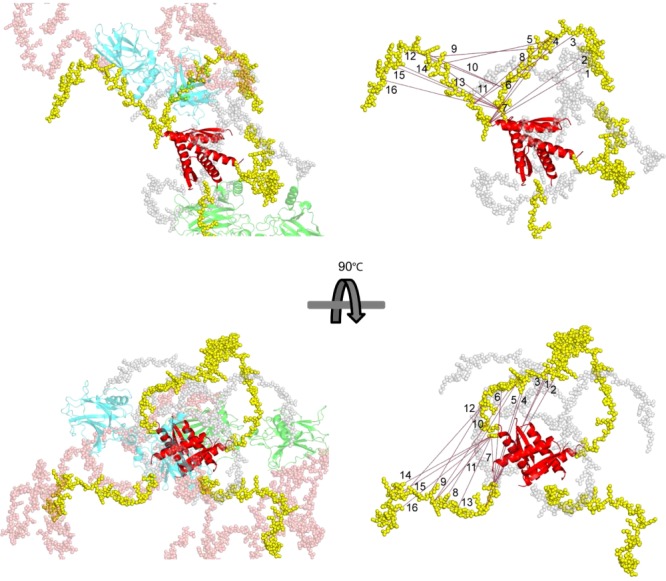
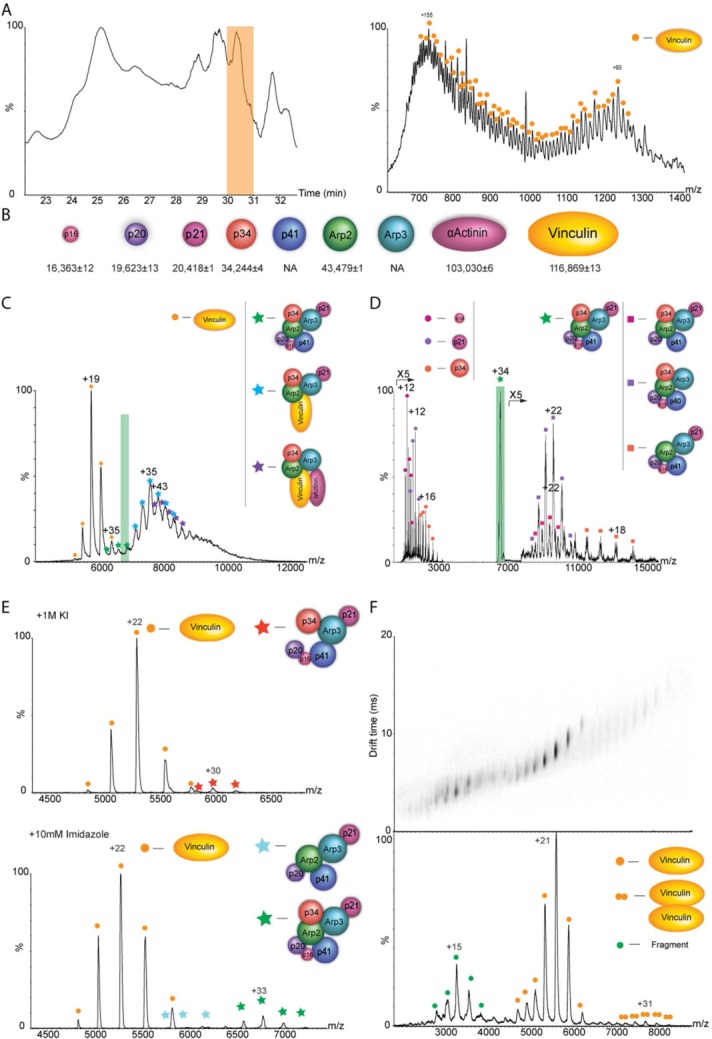
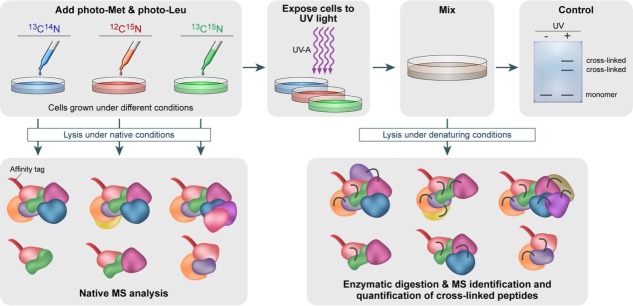
Mass spectrometry (MS) is becoming increasingly popular in the field of structural biology for analyzing protein three-dimensional-structures and for mapping protein-protein interactions. In this review, the specific contributions of chemical crosslinking and native MS are outlined to reveal the structural features of proteins and protein assemblies. Both strategies are illustrated based on the examples of the tetrameric tumor suppressor protein p53 and multisubunit vinculin-Arp2/3 hybrid complexes. We describe the distinct advantages and limitations of each technique and highlight synergistic effects when both techniques are combined. Integrating both methods is especially useful for characterizing large protein assemblies and for capturing transient interactions. We also point out the future directions we foresee for a combination of in vivo crosslinking and native MS for structural investigation of intact protein assemblies.
Keywords: chemical cross-linking; native mass spectrometry; protein 3D structure; protein-protein interactions.
© 2015 The Protein Society.
Figures
References
-
- Tugarinov V, Hwang PM, Kay LE. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev Biochem. 2004;73:107–146. - PubMed
-
- Ilari A, Savino C. Protein structure determination by x-ray crystallography. Methods Mol Biol. 2008;452:63–87. - PubMed
-
- Parrish JR, Gulyas KD, Finley RL., Jr Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol. 2006;17:387–393. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
