

Progress and challenges in predicting protein interfaces
- PMID: 25971595
- PMCID: PMC4719070
- DOI: 10.1093/bib/bbv027
Progress and challenges in predicting protein interfaces
Abstract
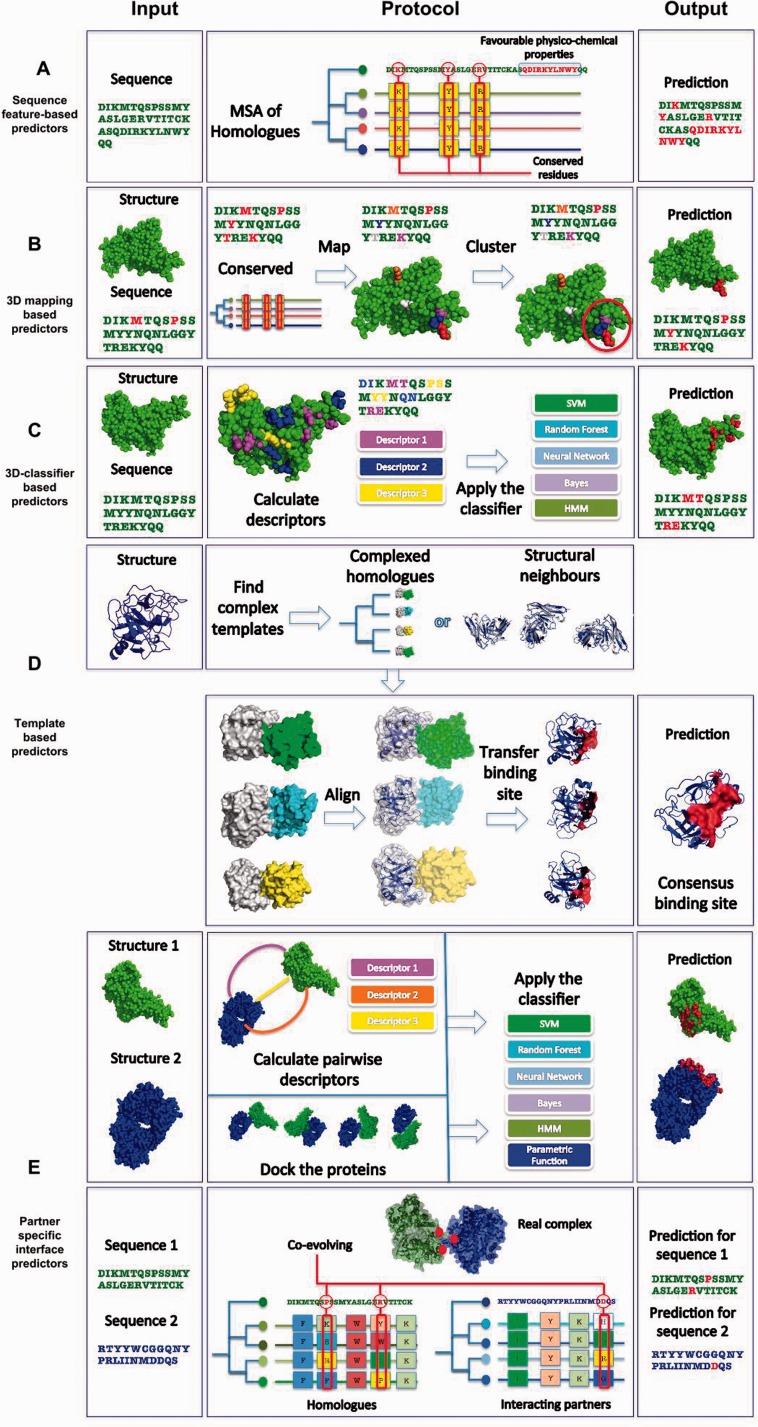
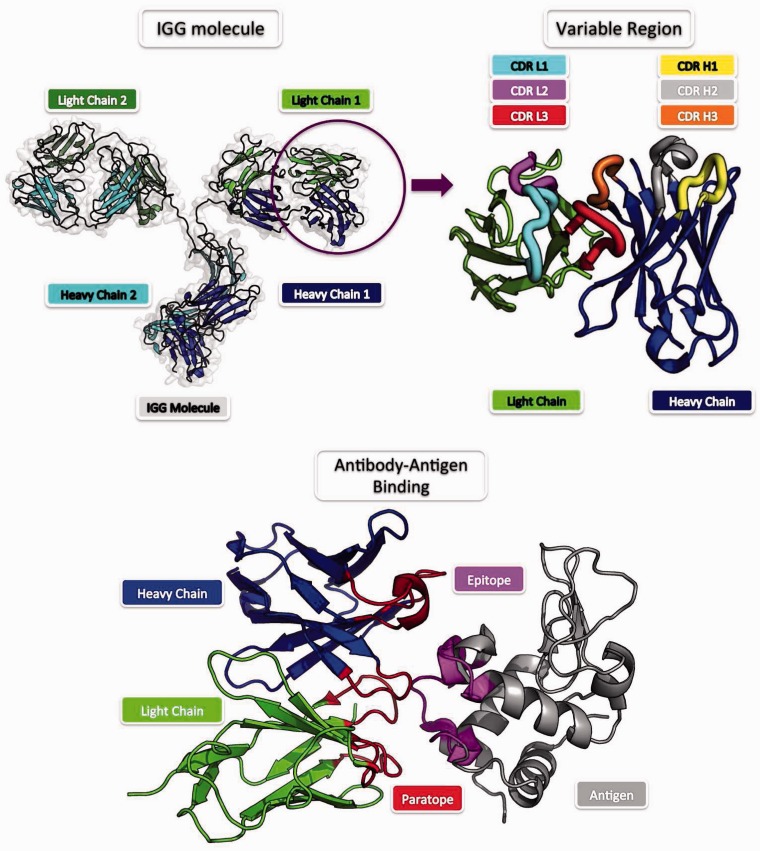
The majority of biological processes are mediated via protein-protein interactions. Determination of residues participating in such interactions improves our understanding of molecular mechanisms and facilitates the development of therapeutics. Experimental approaches to identifying interacting residues, such as mutagenesis, are costly and time-consuming and thus, computational methods for this purpose could streamline conventional pipelines. Here we review the field of computational protein interface prediction. We make a distinction between methods which address proteins in general and those targeted at antibodies, owing to the radically different binding mechanism of antibodies. We organize the multitude of currently available methods hierarchically based on required input and prediction principles to provide an overview of the field.
Keywords: antibody antigen interaction; protein interface prediction; protein–protein interaction.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Sudha G, Nussinov R, Srinivasan N. An overview of recent advances in structural bioinformatics of protein-protein interactions and a guide to their principles. Prog Biophys Mol Biol 2014;116:141–50. - PubMed
-
- Cazals F. Revisiting the Voronoi description of protein-protein interfaces: Algorithms. Pattern Recognit Bioinform 2010;6282:419–30. - PubMed
-
- Janin J, Henrick K, Moult J, et al. CAPRI: a critical assessment of predicted interactions. Proteins 2003;52:2–9. - PubMed
-
- Yan C, Dobbs D, Honavar V. A two-stage classifier for identification of protein-protein interface residues. Bioinformatics 2004;20:i371–8. - PubMed
-
- Ezkurdia I, Bartoli L, Fariselli P, et al. Progress and challenges in predicting protein-protein interaction sites. Brief Bioinform 2009;10:233–46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
