

ISOpureR: an R implementation of a computational purification algorithm of mixed tumour profiles
- PMID: 25972088
- PMCID: PMC4429941
- DOI: 10.1186/s12859-015-0597-x
ISOpureR: an R implementation of a computational purification algorithm of mixed tumour profiles
Abstract
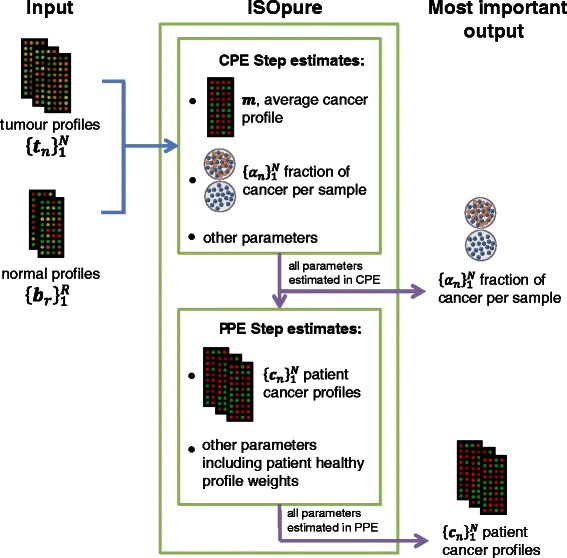
Background: Tumour samples containing distinct sub-populations of cancer and normal cells present challenges in the development of reproducible biomarkers, as these biomarkers are based on bulk signals from mixed tumour profiles. ISOpure is the only mRNA computational purification method to date that does not require a paired tumour-normal sample, provides a personalized cancer profile for each patient, and has been tested on clinical data. Replacing mixed tumour profiles with ISOpure-preprocessed cancer profiles led to better prognostic gene signatures for lung and prostate cancer.
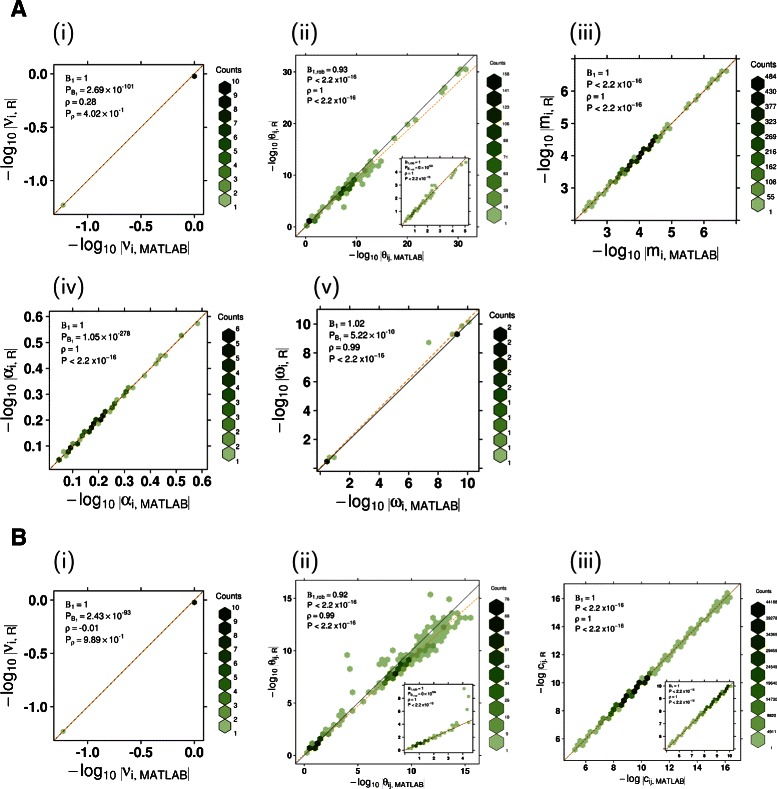
Results: To simplify the integration of ISOpure into standard R-based bioinformatics analysis pipelines, the algorithm has been implemented as an R package. The ISOpureR package performs analogously to the original code in estimating the fraction of cancer cells and the patient cancer mRNA abundance profile from tumour samples in four cancer datasets.
Conclusions: The ISOpureR package estimates the fraction of cancer cells and personalized patient cancer mRNA abundance profile from a mixed tumour profile. This open-source R implementation enables integration into existing computational pipelines, as well as easy testing, modification and extension of the model.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
