

Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation
- PMID: 25972157
- PMCID: PMC4492196
- DOI: 10.1182/blood-2015-02-626846
Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation
Abstract
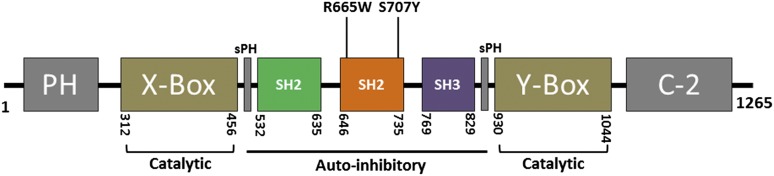
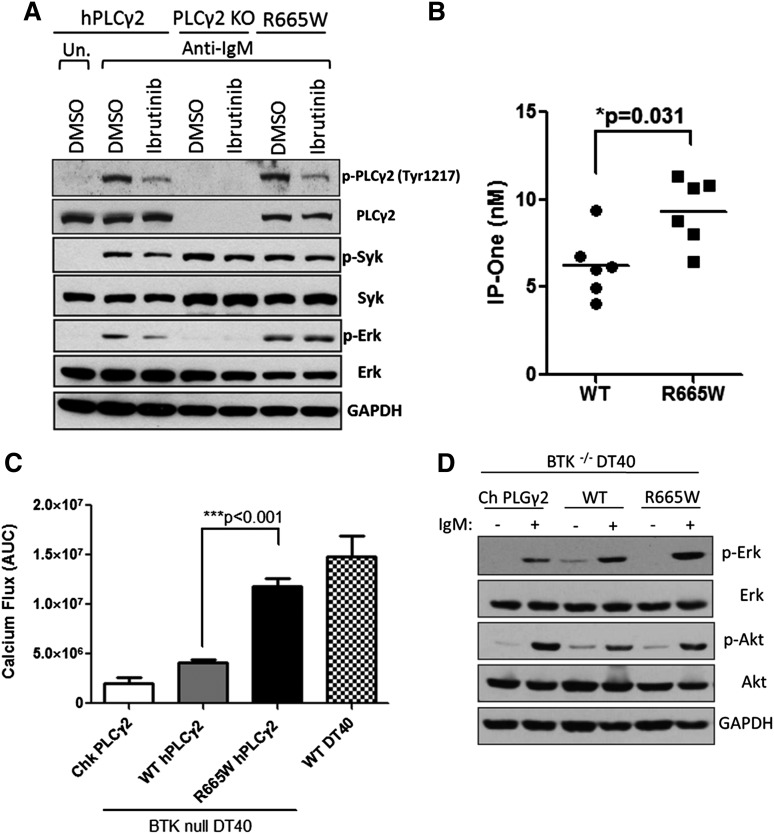
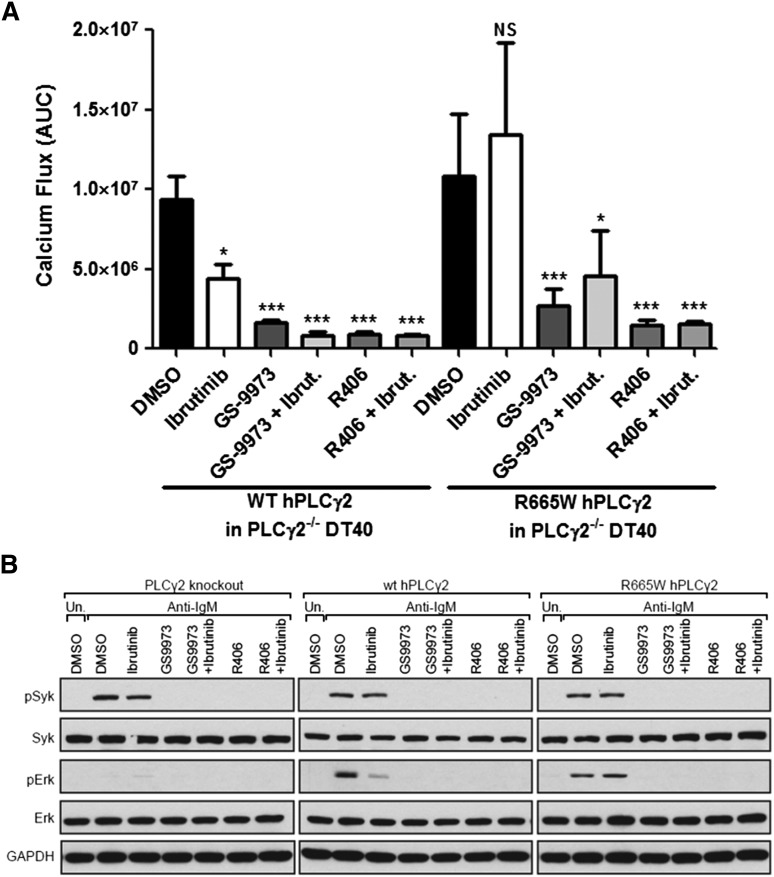
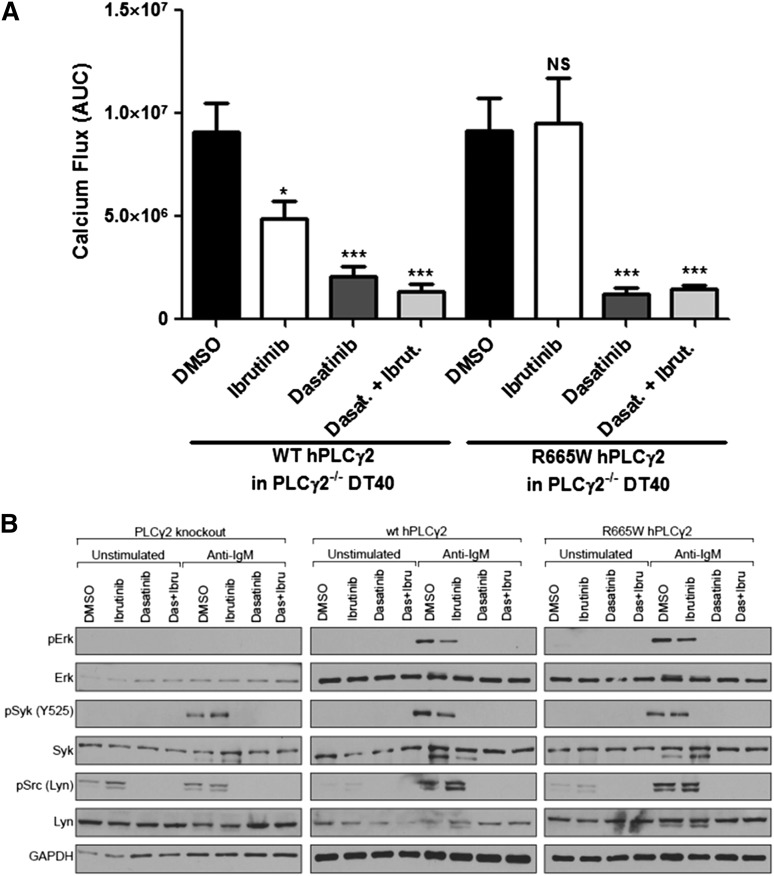
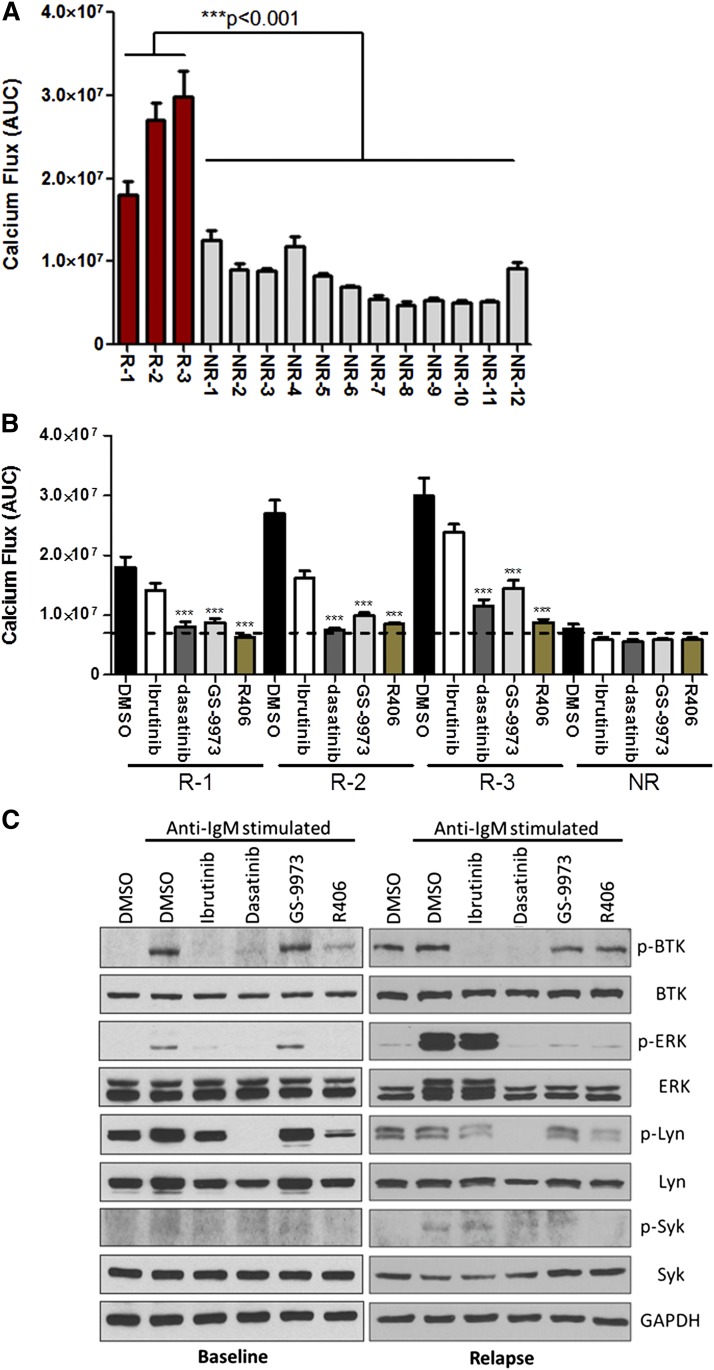
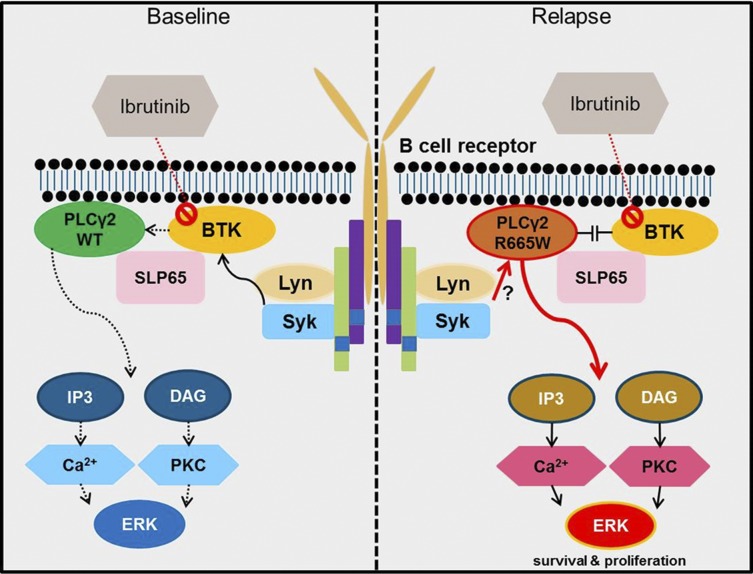
Ibrutinib has significantly improved the outcome of patients with relapsed chronic lymphocytic leukemia (CLL). Recent reports attribute ibrutinib resistance to acquired mutations in Bruton agammaglobulinemia tyrosine kinase (BTK), the target of ibrutinib, as well as the immediate downstream effector phospholipase C, γ2 (PLCG2). Although the C481S mutation found in BTK has been shown to disable ibrutinib's capacity to irreversibly bind this primary target, the detailed mechanisms of mutations in PLCG2 have yet to be established. Herein, we characterize the enhanced signaling competence, BTK independence, and surface immunoglobulin dependence of the PLCG2 mutation at R665W, which has been documented in ibrutinib-resistant CLL. Our data demonstrate that this missense alteration elicits BTK-independent activation after B-cell receptor engagement, implying the formation of a novel BTK-bypass pathway. Consistent with previous results, PLCG2(R665W) confers hypermorphic induction of downstream signaling events. Our studies reveal that proximal kinases SYK and LYN are critical for the activation of mutant PLCG2 and that therapeutics targeting SYK and LYN can combat molecular resistance in cell line models and primary CLL cells from ibrutinib-resistant patients. Altogether, our results engender a molecular understanding of the identified aberration at PLCG2 and explore its functional dependency on BTK, SYK, and LYN, suggesting alternative strategies to combat acquired ibrutinib resistance.
© 2015 by The American Society of Hematology.
Figures
References
-
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. - PubMed
-
- Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103(12):4389–4395. - PubMed
-
- Hallek M, Fischer K, Fingerle-Rowson G, et al. International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
