

Inducing iron deficiency improves erythropoiesis and photosensitivity in congenital erythropoietic porphyria
- PMID: 25972160
- PMCID: PMC4497965
- DOI: 10.1182/blood-2014-07-584664
Inducing iron deficiency improves erythropoiesis and photosensitivity in congenital erythropoietic porphyria
Abstract
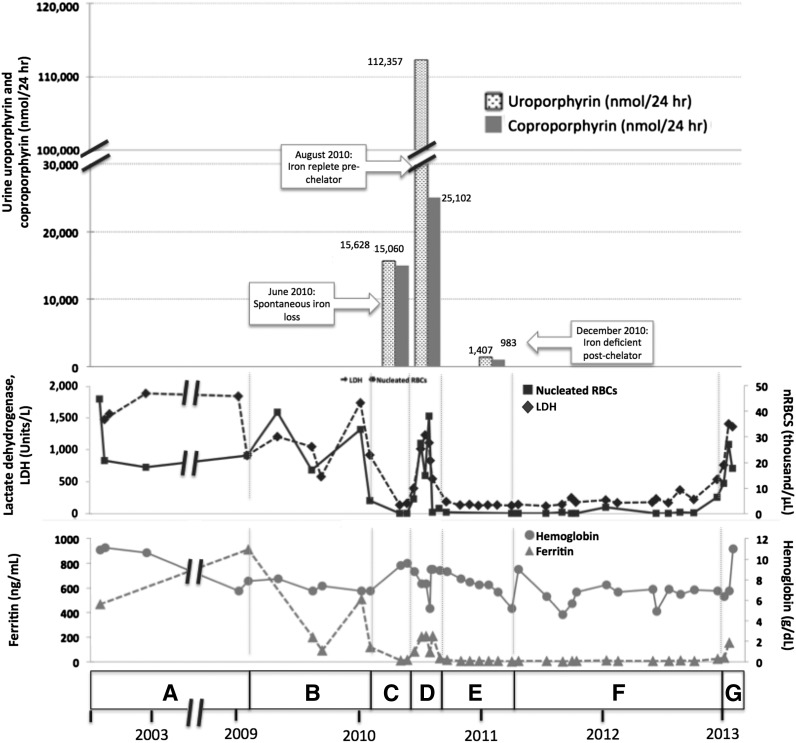
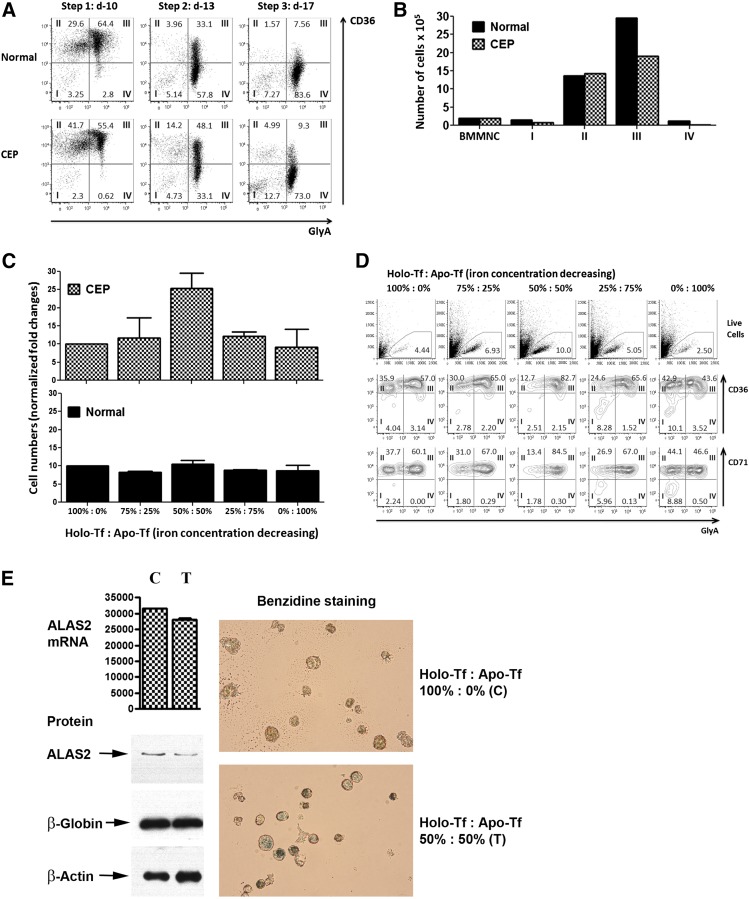
Congenital erythropoietic porphyria (CEP) is an autosomal recessive disorder of heme synthesis characterized by reduced activity of uroporphyrinogen III synthase and the accumulation of nonphysiologic isomer I porphyrin metabolites, resulting in ineffective erythropoiesis and devastating skin photosensitivity. Management of the disease primarily consists of supportive measures. Increased activity of 5-aminolevulinate synthase 2 (ALAS2) has been shown to adversely modify the disease phenotype. Herein, we present a patient with CEP who demonstrated a remarkable improvement in disease manifestations in the setting of iron deficiency. Hypothesizing that iron restriction improved her symptoms by decreasing ALAS2 activity and subsequent porphyrin production, we treated the patient with off-label use of deferasirox to maintain iron deficiency, with successful results. We confirmed the physiology of her response with marrow culture studies.
© 2015 by The American Society of Hematology.
Figures
Comment in
-
Iron and erythropoietic porphyrias.Blood. 2015 Jul 9;126(2):130-2. doi: 10.1182/blood-2015-05-646737. Blood. 2015. PMID: 26160187 No abstract available.
References
-
- Thomas C, Ged C, Nordmann Y, et al. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr. 1996;129(3):453–456. - PubMed
-
- Zix-Kieffer I, Langer B, Eyer D, et al. Successful cord blood stem cell transplantation for congenital erythropoietic porphyria (Gunther’s disease). Bone Marrow Transplant. 1996;18(1):217–220. - PubMed
-
- Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol. 2001;45(2):279–282. - PubMed
-
- Ged C, Moreau-Gaudry F, Richard E, Robert-Richard E, de Verneuil H. Congenital erythropoietic porphyria: mutation update and correlations between genotype and phenotype. Cell Mol Biol (Noisy-le-grand) 2009;55(1):53–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
