

Dominant, toxic gain-of-function mutations in gars lead to non-cell autonomous neuropathology
- PMID: 25972375
- PMCID: PMC4492401
- DOI: 10.1093/hmg/ddv176
Dominant, toxic gain-of-function mutations in gars lead to non-cell autonomous neuropathology
Abstract
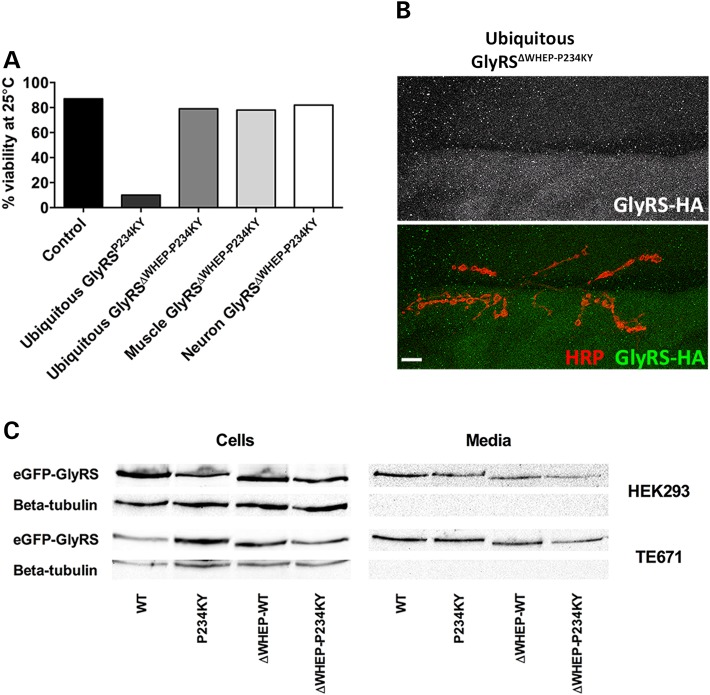
Charcot-Marie-Tooth (CMT) neuropathies are collectively the most common hereditary neurological condition and a major health burden for society. Dominant mutations in the gene GARS, encoding the ubiquitous enzyme, glycyl-tRNA synthetase (GlyRS), cause peripheral nerve degeneration and lead to CMT disease type 2D. This genetic disorder exemplifies a recurring motif in neurodegeneration, whereby mutations in essential, widely expressed genes have selective deleterious consequences for the nervous system. Here, using novel Drosophila models, we show a potential solution to this phenomenon. Ubiquitous expression of mutant GlyRS leads to motor deficits, progressive neuromuscular junction (NMJ) denervation and pre-synaptic build-up of mutant GlyRS. Intriguingly, neuronal toxicity is, at least in part, non-cell autonomous, as expression of mutant GlyRS in mesoderm or muscle alone results in similar pathology. This mutant GlyRS toxic gain-of-function, which is WHEP domain-dependent, coincides with abnormal NMJ assembly, leading to synaptic degeneration, and, ultimately, reduced viability. Our findings suggest that mutant GlyRS gains access to ectopic sub-compartments of the motor neuron, providing a possible explanation for the selective neuropathology caused by mutations in a widely expressed gene.
© The Author 2015. Published by Oxford University Press.
Figures




References
-
- Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T., et al. (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. - PMC - PubMed
-
- Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V., et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet., 38, 197–202. - PubMed
-
- Latour P., Thauvin-Robinet C., Baudelet-Mery C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayencon M., Sarcey A., Broussolle E., et al. (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet., 86, 77–82. - PMC - PubMed
-
- Gonzalez M., McLaughlin H., Houlden H., Guo M., Yo-Tsen L., Hadjivassilious M., Speziani F., Yang X.L., Antonellis A., Reilly M.M., et al. (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry, 84, 1247–1249. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
