

High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination
- PMID: 25972543
- PMCID: PMC4505652
- DOI: 10.1128/JVI.00578-15
High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination
Abstract
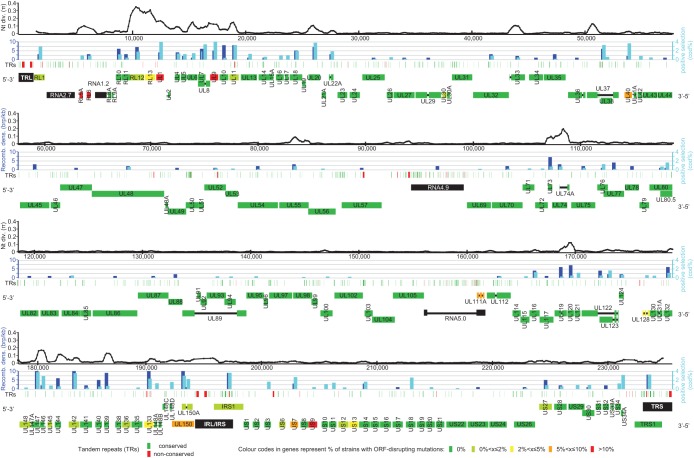
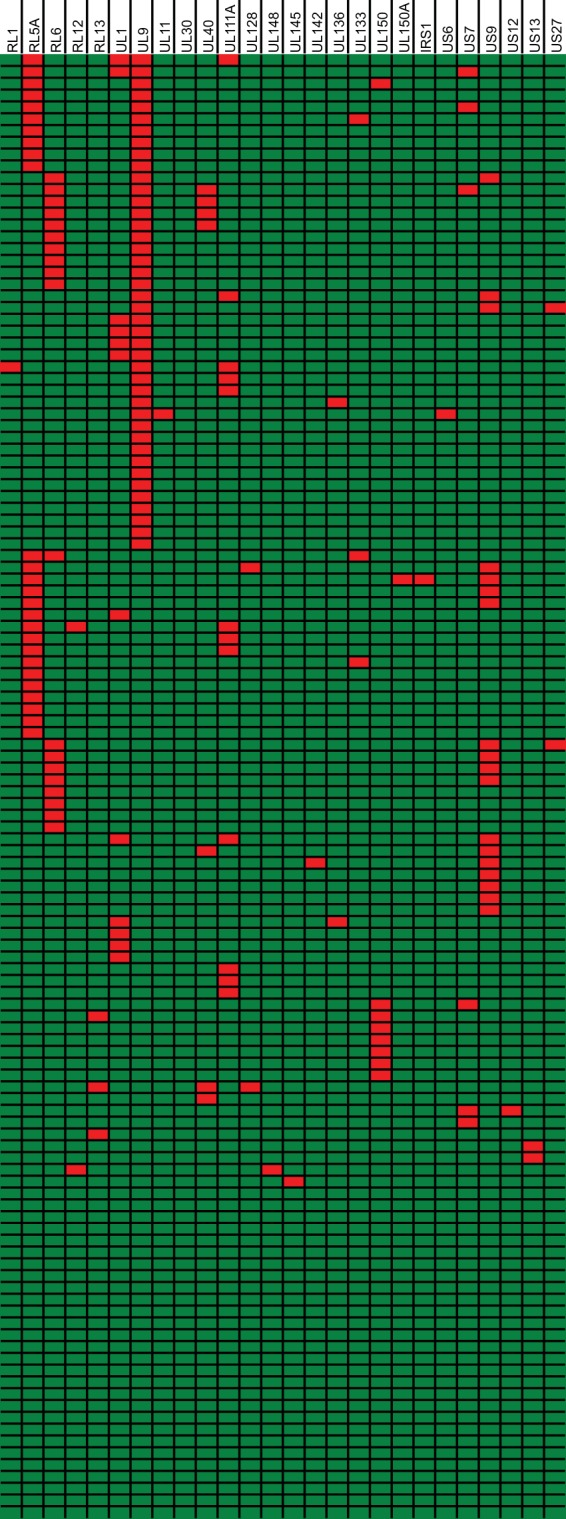
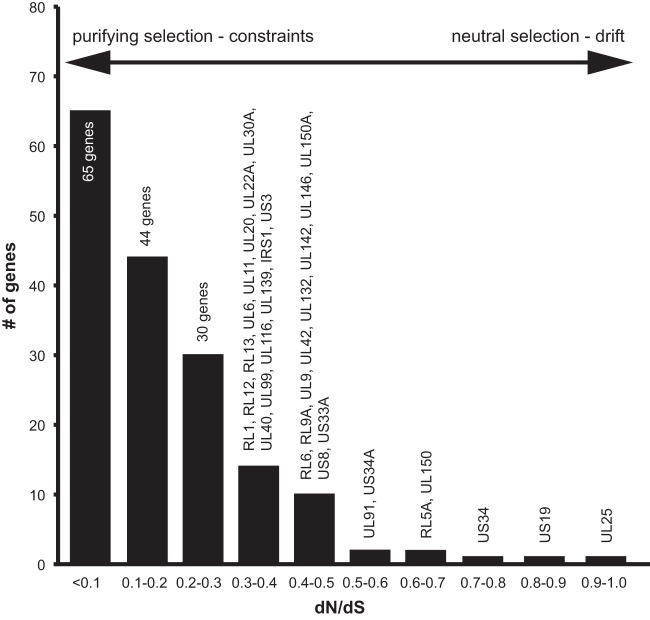
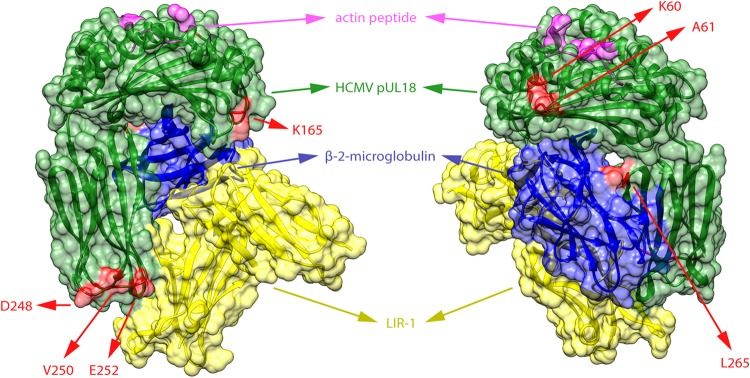
Human cytomegalovirus is a widespread pathogen of major medical importance. It causes significant morbidity and mortality in the immunocompromised and congenital infections can result in severe disabilities or stillbirth. Development of a vaccine is prioritized, but no candidate is close to release. Although correlations of viral genetic variability with pathogenicity are suspected, knowledge about strain diversity of the 235kb genome is still limited. In this study, 96 full-length human cytomegalovirus genomes from clinical isolates were characterized, quadrupling the available information for full-genome analysis. These data provide the first high-resolution map of human cytomegalovirus interhost diversity and evolution. We show that cytomegalovirus is significantly more divergent than all other human herpesviruses and highlight hotspots of diversity in the genome. Importantly, 75% of strains are not genetically intact, but contain disruptive mutations in a diverse set of 26 genes, including immunomodulative genes UL40 and UL111A. These mutants are independent from culture passaging artifacts and circulate in natural populations. Pervasive recombination, which is linked to the widespread occurrence of multiple infections, was found throughout the genome. Recombination density was significantly higher than in other human herpesviruses and correlated with strain diversity. While the overall effects of strong purifying selection on virus evolution are apparent, evidence of diversifying selection was found in several genes encoding proteins that interact with the host immune system, including UL18, UL40, UL142 and UL147. These residues may present phylogenetic signatures of past and ongoing virus-host interactions.
Importance: Human cytomegalovirus has the largest genome of all viruses that infect humans. Currently, there is a great interest in establishing associations between genetic variants and strain pathogenicity of this herpesvirus. Since the number of publicly available full-genome sequences is limited, knowledge about strain diversity is highly fragmented and biased towards a small set of loci. Combined with our previous work, we have now contributed 101 complete genome sequences. We have used these data to conduct the first high-resolution analysis of interhost genome diversity, providing an unbiased and comprehensive overview of cytomegalovirus variability. These data are of major value to the development of novel antivirals and a vaccine and to identify potential targets for genotype-phenotype experiments. Furthermore, they have enabled a thorough study of the evolutionary processes that have shaped cytomegalovirus diversity.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures





References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
