

Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury during Pneumococcal Infection
- PMID: 25973949
- PMCID: PMC4431880
- DOI: 10.1371/journal.ppat.1004836
Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury during Pneumococcal Infection
Abstract
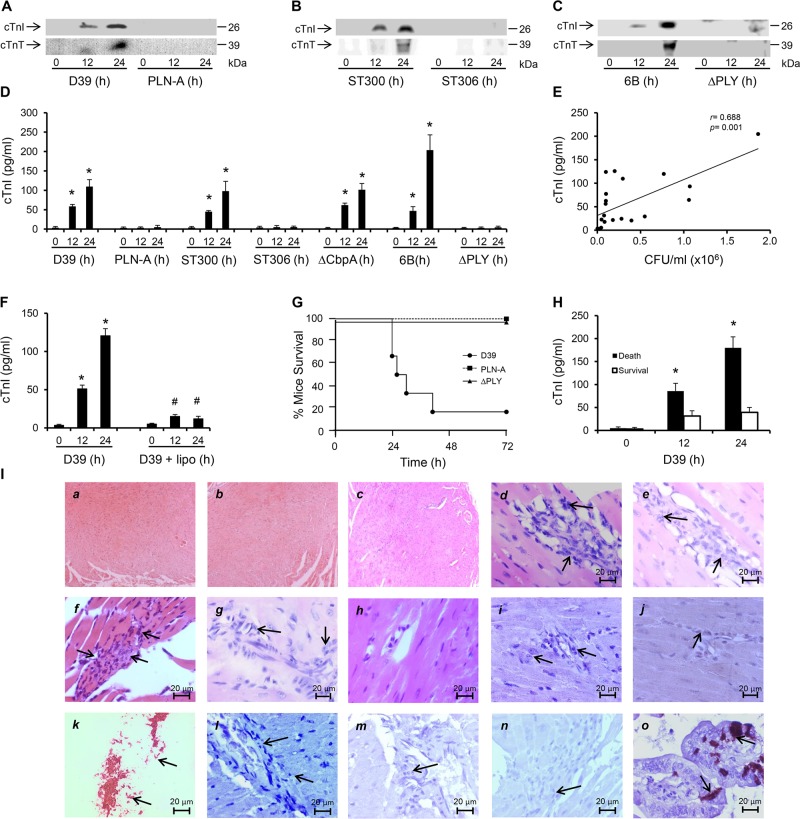
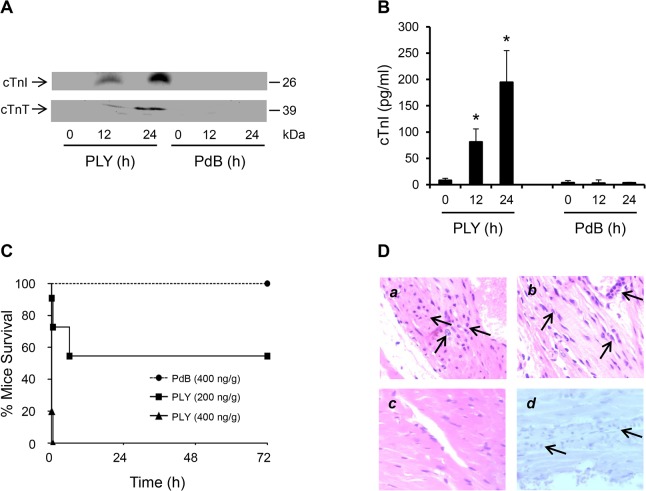
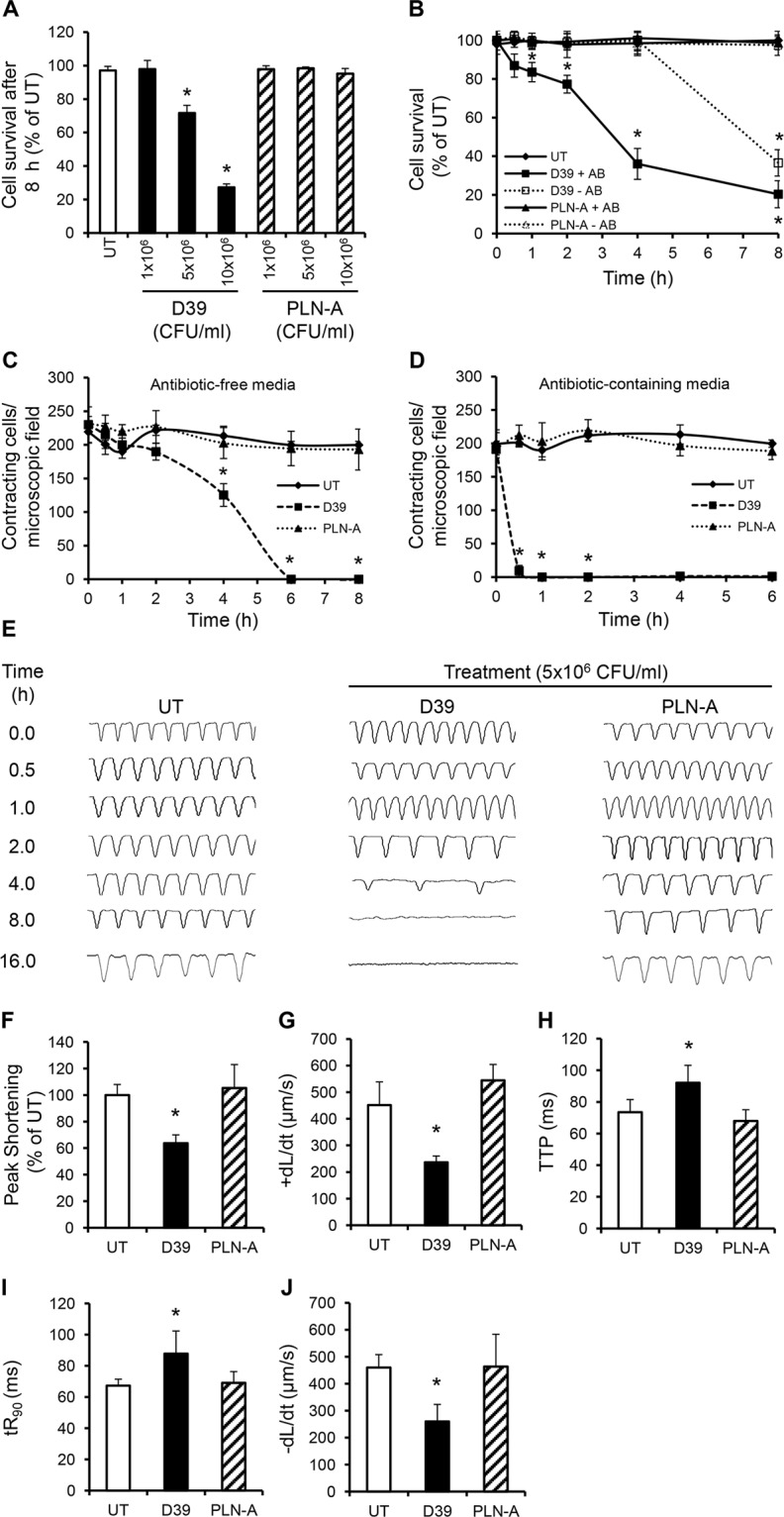
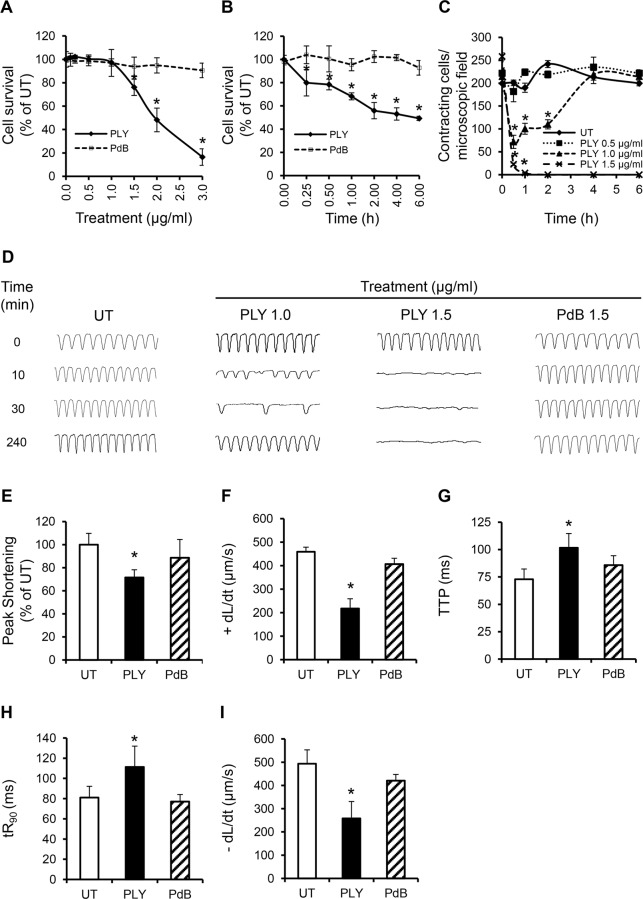
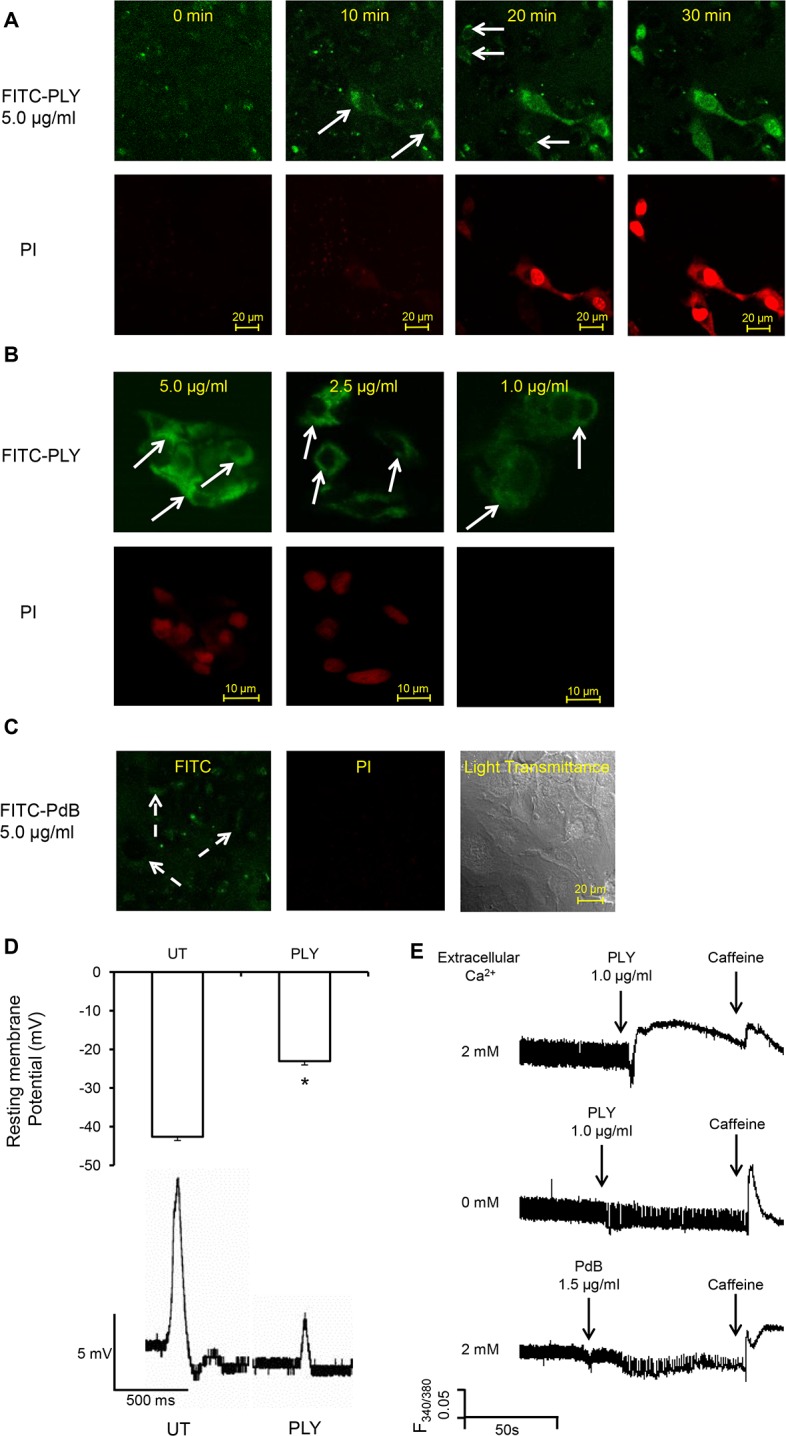
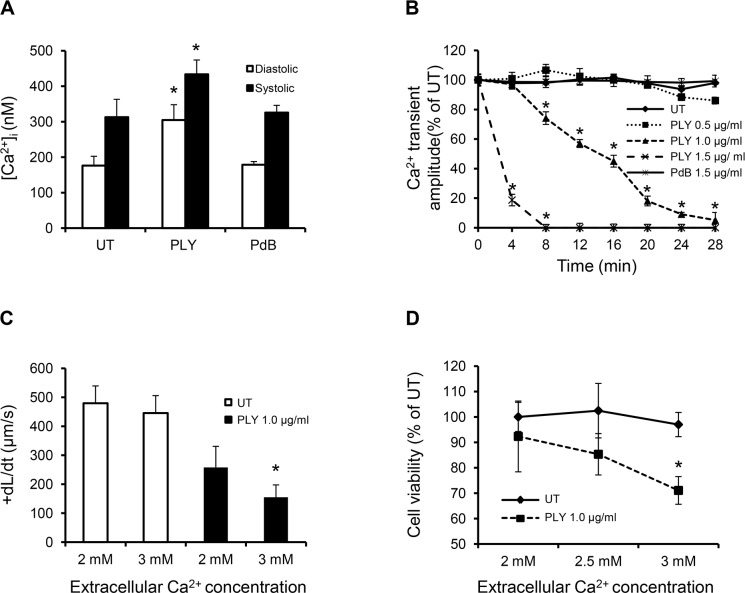
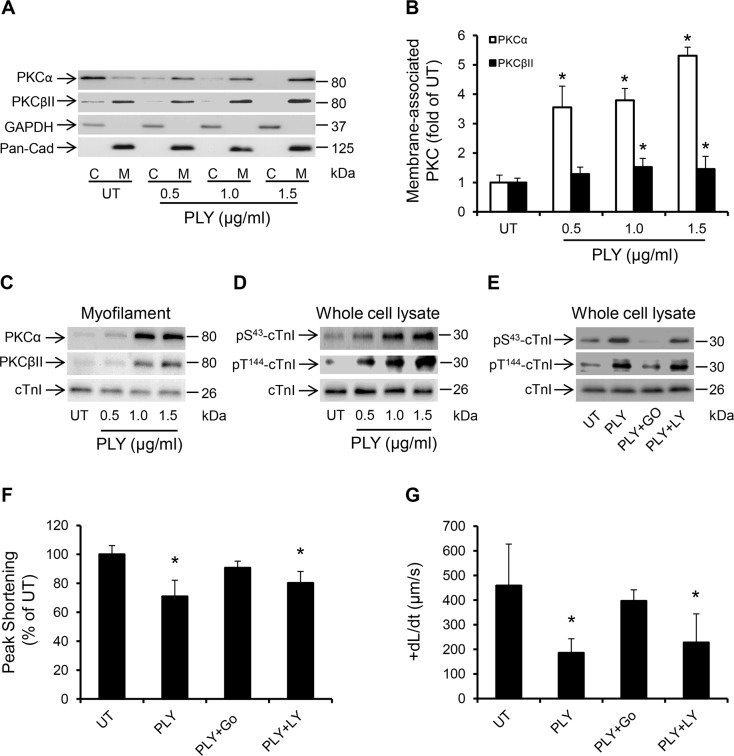
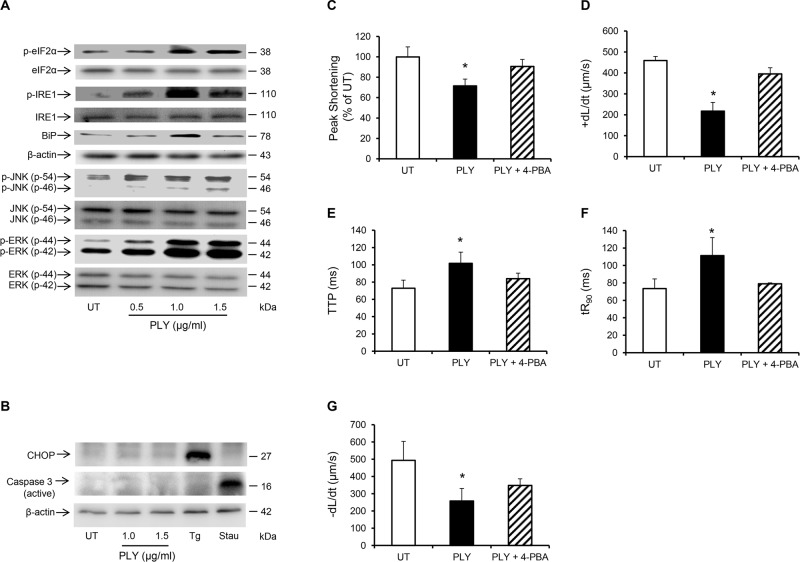
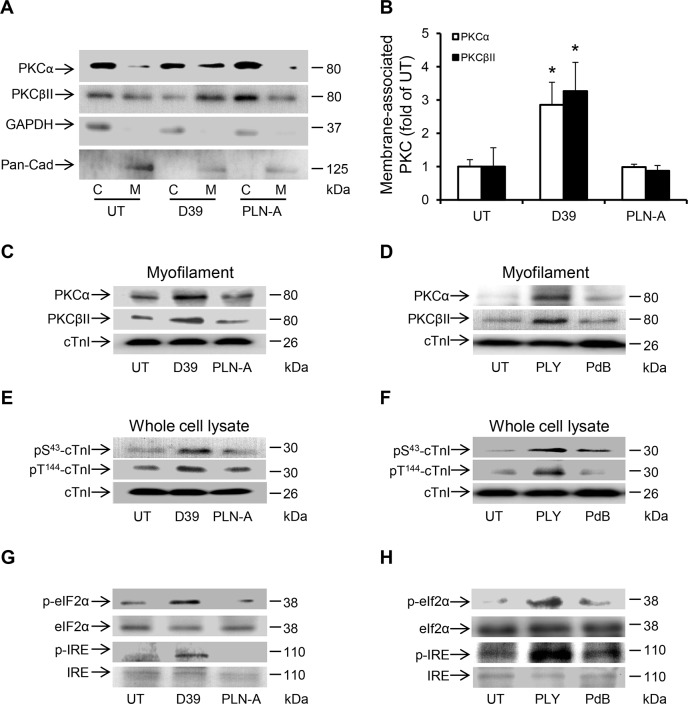
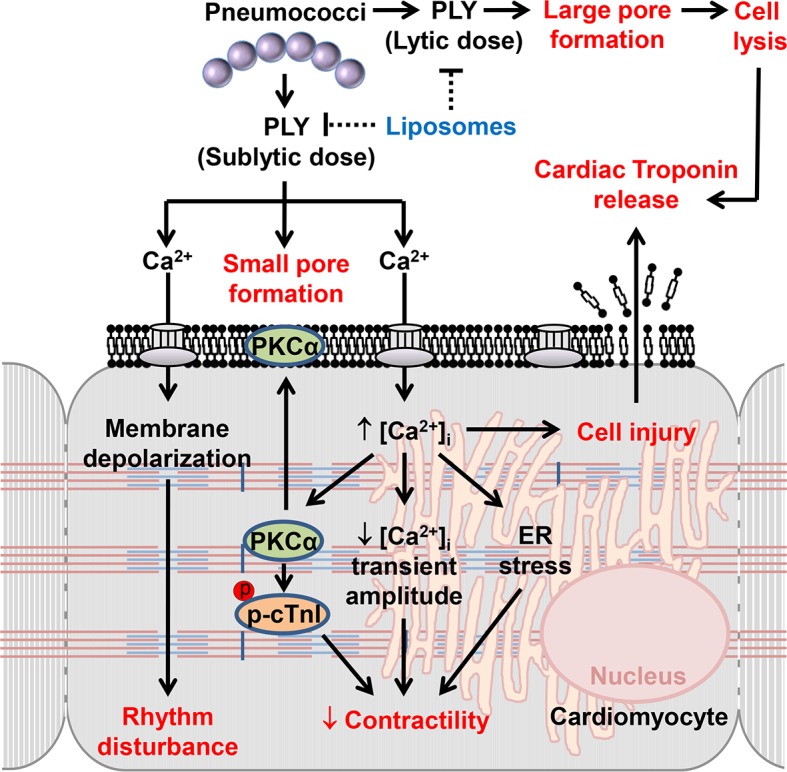
Streptococcus pneumoniae accounts for more deaths worldwide than any other single pathogen through diverse disease manifestations including pneumonia, sepsis and meningitis. Life-threatening acute cardiac complications are more common in pneumococcal infection compared to other bacterial infections. Distinctively, these arise despite effective antibiotic therapy. Here, we describe a novel mechanism of myocardial injury, which is triggered and sustained by circulating pneumolysin (PLY). Using a mouse model of invasive pneumococcal disease (IPD), we demonstrate that wild type PLY-expressing pneumococci but not PLY-deficient mutants induced elevation of circulating cardiac troponins (cTns), well-recognized biomarkers of cardiac injury. Furthermore, elevated cTn levels linearly correlated with pneumococcal blood counts (r=0.688, p=0.001) and levels were significantly higher in non-surviving than in surviving mice. These cTn levels were significantly reduced by administration of PLY-sequestering liposomes. Intravenous injection of purified PLY, but not a non-pore forming mutant (PdB), induced substantial increase in cardiac troponins to suggest that the pore-forming activity of circulating PLY is essential for myocardial injury in vivo. Purified PLY and PLY-expressing pneumococci also caused myocardial inflammatory changes but apoptosis was not detected. Exposure of cultured cardiomyocytes to PLY-expressing pneumococci caused dose-dependent cardiomyocyte contractile dysfunction and death, which was exacerbated by further PLY release following antibiotic treatment. We found that high PLY doses induced extensive cardiomyocyte lysis, but more interestingly, sub-lytic PLY concentrations triggered profound calcium influx and overload with subsequent membrane depolarization and progressive reduction in intracellular calcium transient amplitude, a key determinant of contractile force. This was coupled to activation of signalling pathways commonly associated with cardiac dysfunction in clinical and experimental sepsis and ultimately resulted in depressed cardiomyocyte contractile performance along with rhythm disturbance. Our study proposes a detailed molecular mechanism of pneumococcal toxin-induced cardiac injury and highlights the major translational potential of targeting circulating PLY to protect against cardiac complications during pneumococcal infections.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Ortqvist A, Hedlund J, Kalin M (2005) Streptococcus pneumoniae: epidemiology, risk factors, and clinical features. Semin Respir Crit Care Med 26: 563–574. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
