

CONFOLD: Residue-residue contact-guided ab initio protein folding
- PMID: 25974172
- PMCID: PMC4509844
- DOI: 10.1002/prot.24829
CONFOLD: Residue-residue contact-guided ab initio protein folding
Abstract
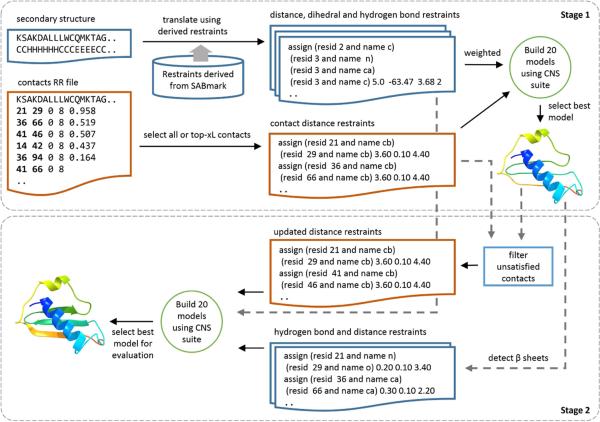
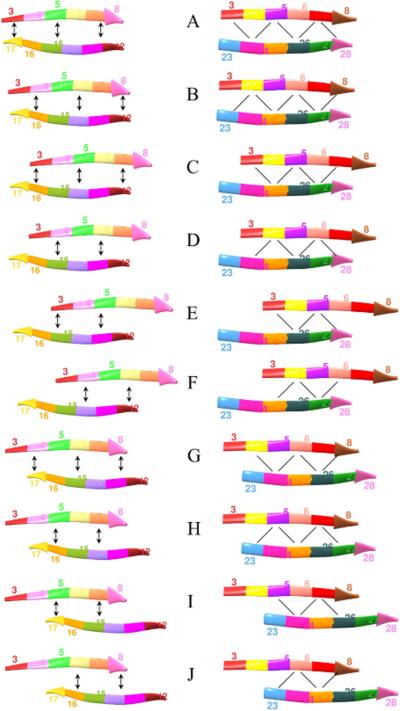
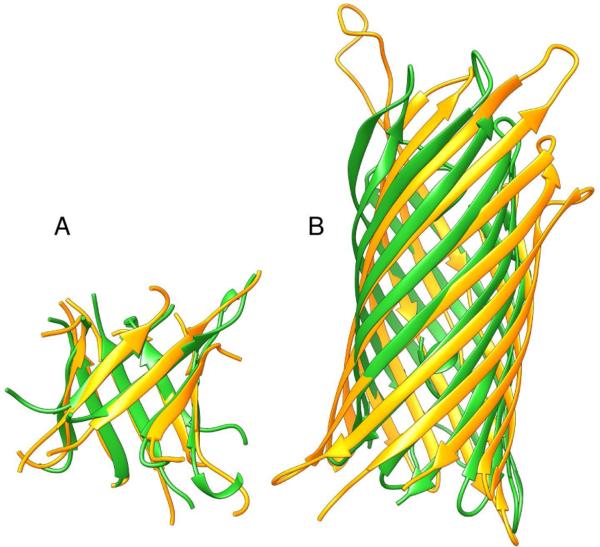
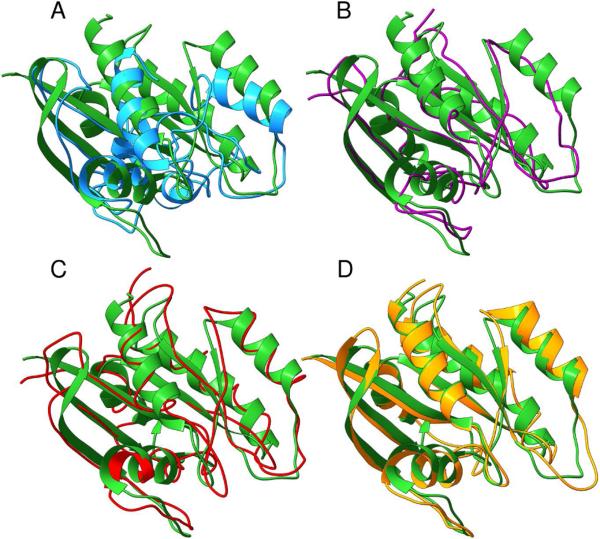
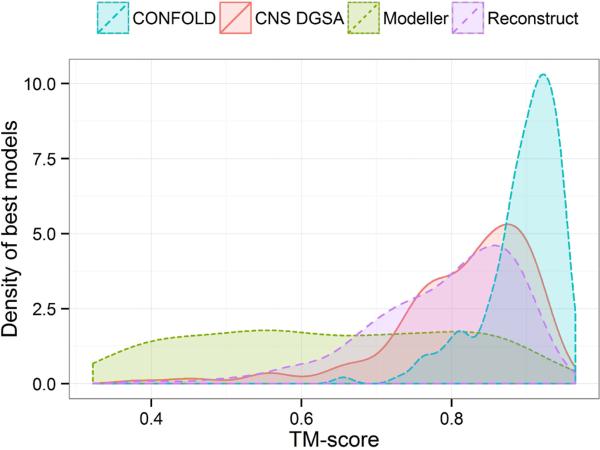
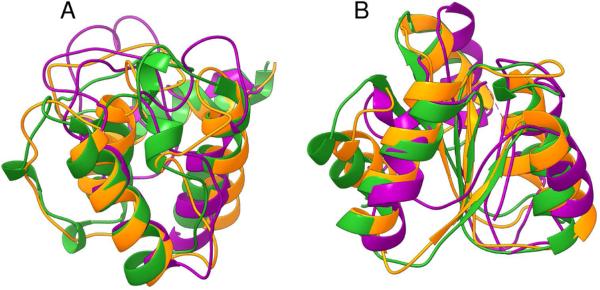
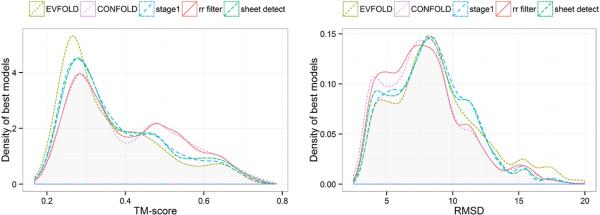
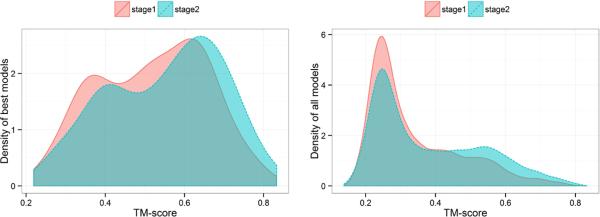
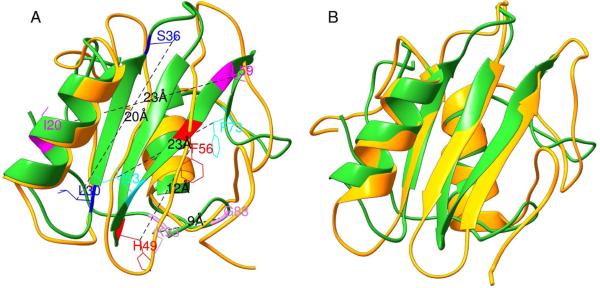
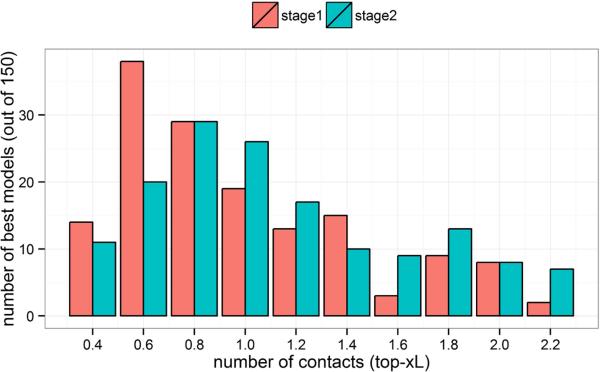
Predicted protein residue-residue contacts can be used to build three-dimensional models and consequently to predict protein folds from scratch. A considerable amount of effort is currently being spent to improve contact prediction accuracy, whereas few methods are available to construct protein tertiary structures from predicted contacts. Here, we present an ab initio protein folding method to build three-dimensional models using predicted contacts and secondary structures. Our method first translates contacts and secondary structures into distance, dihedral angle, and hydrogen bond restraints according to a set of new conversion rules, and then provides these restraints as input for a distance geometry algorithm to build tertiary structure models. The initially reconstructed models are used to regenerate a set of physically realistic contact restraints and detect secondary structure patterns, which are then used to reconstruct final structural models. This unique two-stage modeling approach of integrating contacts and secondary structures improves the quality and accuracy of structural models and in particular generates better β-sheets than other algorithms. We validate our method on two standard benchmark datasets using true contacts and secondary structures. Our method improves TM-score of reconstructed protein models by 45% and 42% over the existing method on the two datasets, respectively. On the dataset for benchmarking reconstructions methods with predicted contacts and secondary structures, the average TM-score of best models reconstructed by our method is 0.59, 5.5% higher than the existing method. The CONFOLD web server is available at http://protein.rnet.missouri.edu/confold/.
Keywords: ab initio protein folding; contact assisted protein structure prediction; optimization; protein residue-residue contacts; protein structure modeling.
© 2015 Wiley Periodicals, Inc.
Figures
References
-
- Fariselli P, Olmea O, Valencia A, Casadio R. Prediction of contact maps with neural networks and correlated mutations. Protein engineering. 2001;14(11):835–843. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
