

A repository of assays to quantify 10,000 human proteins by SWATH-MS
- PMID: 25977788
- PMCID: PMC4322573
- DOI: 10.1038/sdata.2014.31
A repository of assays to quantify 10,000 human proteins by SWATH-MS
Abstract
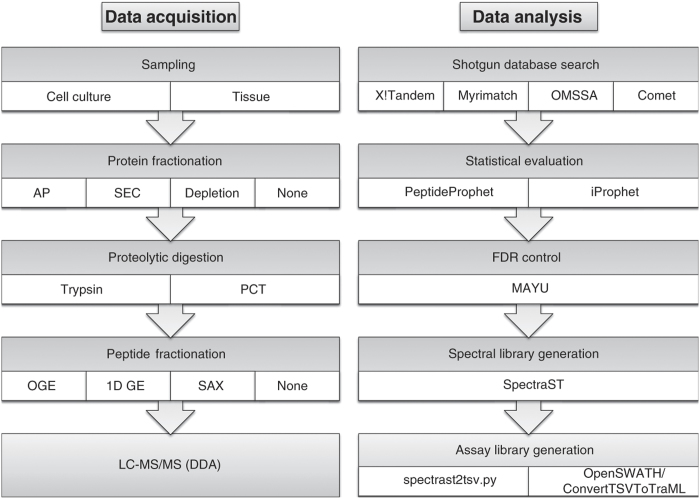
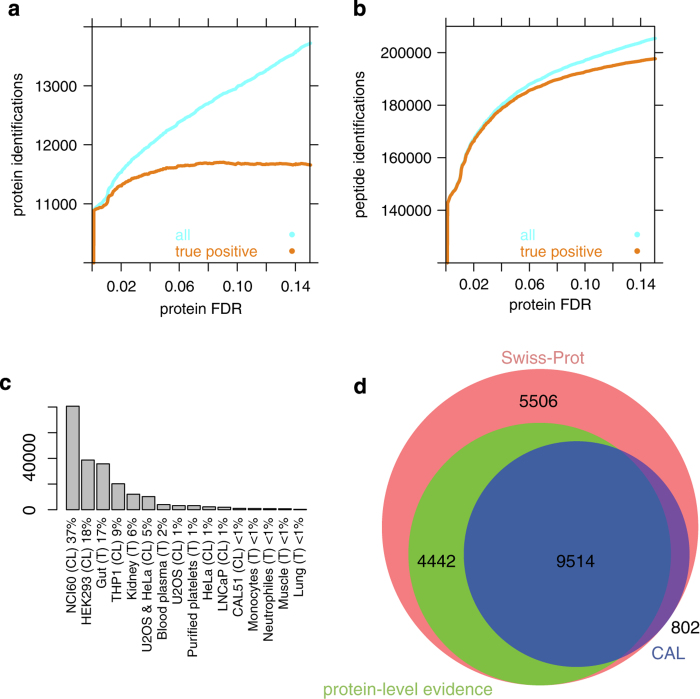
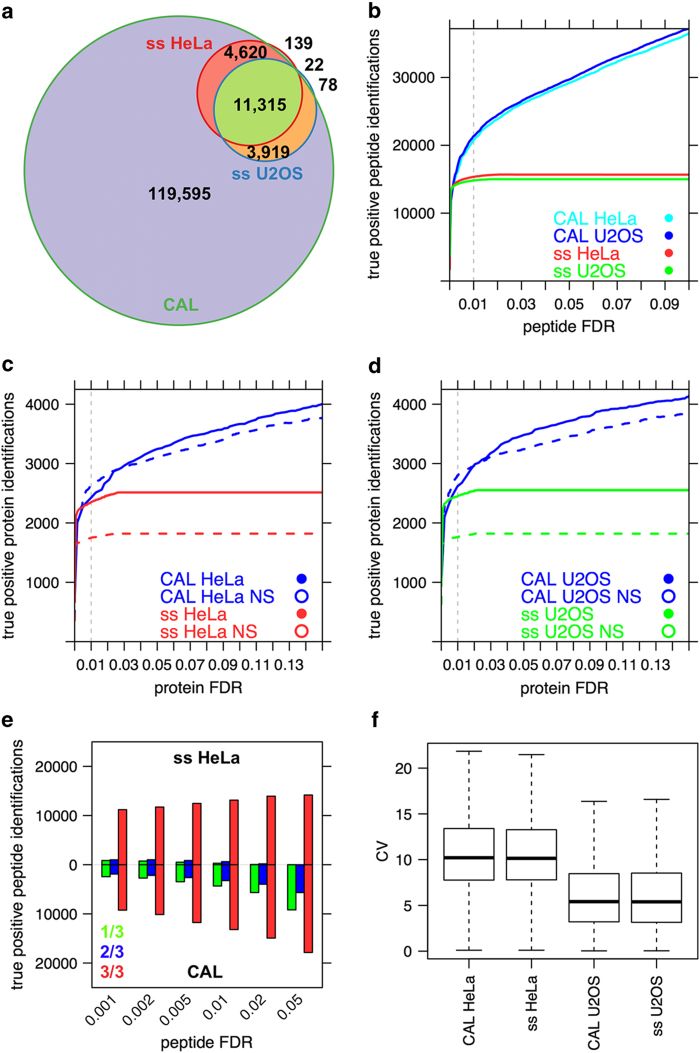
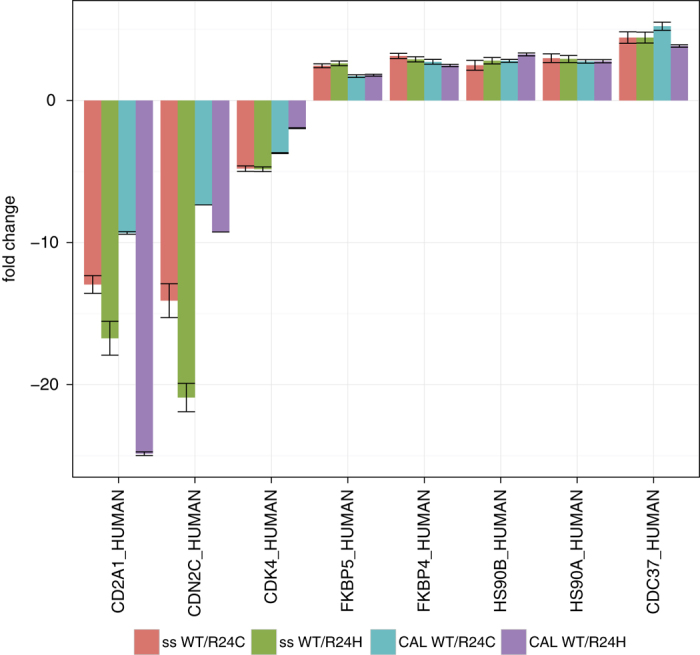
Mass spectrometry is the method of choice for deep and reliable exploration of the (human) proteome. Targeted mass spectrometry reliably detects and quantifies pre-determined sets of proteins in a complex biological matrix and is used in studies that rely on the quantitatively accurate and reproducible measurement of proteins across multiple samples. It requires the one-time, a priori generation of a specific measurement assay for each targeted protein. SWATH-MS is a mass spectrometric method that combines data-independent acquisition (DIA) and targeted data analysis and vastly extends the throughput of proteins that can be targeted in a sample compared to selected reaction monitoring (SRM). Here we present a compendium of highly specific assays covering more than 10,000 human proteins and enabling their targeted analysis in SWATH-MS datasets acquired from research or clinical specimens. This resource supports the confident detection and quantification of 50.9% of all human proteins annotated by UniProtKB/Swiss-Prot and is therefore expected to find wide application in basic and clinical research. Data are available via ProteomeXchange (PXD000953-954) and SWATHAtlas (SAL00016-35).
Conflict of interest statement
S.T. is employee of AB SCIEX, which operates in the field covered by the article. The research group of R.A. is supported in part by AB SCIEX by providing access to prototype instrumentation. R.A. holds shares of Biognosys AG, which operates in the field covered by the article. The remaining authors declare no competing financial interest.
Figures
References
Data Citations
-
- Rosenberger G. 2014. ProteomeXchange. PXD000953
-
- Rosenberger G. 2014. SWATHAtlas. SAL00016-35
-
- Rosenberger G. 2014. ProteomeXchange. PXD000954
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
