

Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures
- PMID: 25979079
- PMCID: PMC4504977
- DOI: 10.1152/ajplung.00411.2014
Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures
Abstract
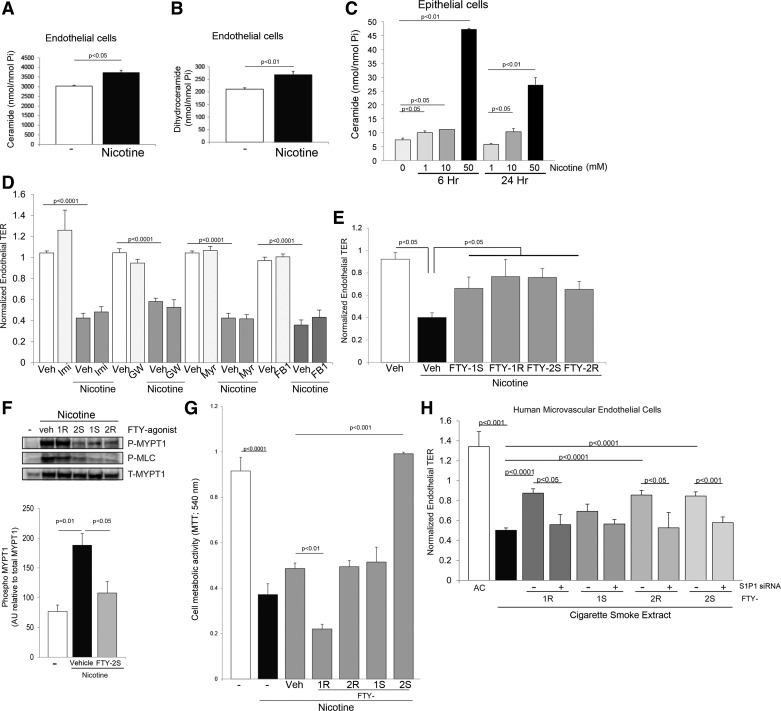
The increased use of inhaled nicotine via e-cigarettes has unknown risks to lung health. Having previously shown that cigarette smoke (CS) extract disrupts the lung microvasculature barrier function by endothelial cell activation and cytoskeletal rearrangement, we investigated the contribution of nicotine in CS or e-cigarettes (e-Cig) to lung endothelial injury. Primary lung microvascular endothelial cells were exposed to nicotine, e-Cig solution, or condensed e-Cig vapor (1-20 mM nicotine) or to nicotine-free CS extract or e-Cig solutions. Compared with nicotine-containing extract, nicotine free-CS extract (10-20%) caused significantly less endothelial permeability as measured with electric cell-substrate impedance sensing. Nicotine exposures triggered dose-dependent loss of endothelial barrier in cultured cell monolayers and rapidly increased lung inflammation and oxidative stress in mice. The endothelial barrier disruptive effects were associated with increased intracellular ceramides, p38 MAPK activation, and myosin light chain (MLC) phosphorylation, and was critically mediated by Rho-activated kinase via inhibition of MLC-phosphatase unit MYPT1. Although nicotine at sufficient concentrations to cause endothelial barrier loss did not trigger cell necrosis, it markedly inhibited cell proliferation. Augmentation of sphingosine-1-phosphate (S1P) signaling via S1P1 improved both endothelial cell proliferation and barrier function during nicotine exposures. Nicotine-independent effects of e-Cig solutions were noted, which may be attributable to acrolein, detected along with propylene glycol, glycerol, and nicotine by NMR, mass spectrometry, and gas chromatography, in both e-Cig solutions and vapor. These results suggest that soluble components of e-Cig, including nicotine, cause dose-dependent loss of lung endothelial barrier function, which is associated with oxidative stress and brisk inflammation.
Keywords: cell proliferation; inflammation; permeability; sphingosine-1-phosphate; tobacco.
Figures







References
-
- Beckers CM, Knezevic N, Valent ET, Tauseef M, Krishnan R, Rajendran K, Corey Hardin C, Aman J, van Bezu J, Sweetnam P, van Hinsbergh VW, Mehta D, van Nieuw Amerongen GP. ROCK2 primes the endothelium for vascular hyperpermeability responses by raising baseline junctional tension. Vascul Pharmacol. First published April 11, 2015; doi: 10.1016/j.vph.2015..03.017. - DOI - PMC - PubMed
-
- Brown MB, Hunt WR, Noe JE, Rush NI, Schweitzer KS, Leece TC, Moldobaeva A, Wagner EM, Dudek SM, Poirier C, Presson RG Jr, Gulbins E, Petrache I. Loss of cystic fibrosis transmembrane conductance regulator impairs lung endothelial cell barrier function and increases susceptibility to microvascular damage from cigarette smoke. Pulm Circ 4: 260–268, 2014. - PMC - PubMed
-
- Cheadle GA, Costantini TW, Bansal V, Eliceiri BP, Coimbra R. Cholinergic signaling in the gut: a novel mechanism of barrier protection through activation of enteric glia cells. Surg Infect (Larchmt) 15: 387–393, 2014. - PubMed
-
- Diab KJ, Adamowicz JJ, Kamocki K, Rush NI, Garrison J, Gu Y, Schweitzer KS, Skobeleva A, Rajashekhar G, Hubbard WC, Berdyshev EV, Petrache I. Stimulation of sphingosine 1-phosphate signaling as an alveolar cell survival strategy in emphysema. Am J Respir Crit Care Med 181: 344–352, 2010. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical