

A microfibril assembly assay identifies different mechanisms of dominance underlying Marfan syndrome, stiff skin syndrome and acromelic dysplasias
- PMID: 25979247
- PMCID: PMC4492404
- DOI: 10.1093/hmg/ddv181
A microfibril assembly assay identifies different mechanisms of dominance underlying Marfan syndrome, stiff skin syndrome and acromelic dysplasias
Abstract
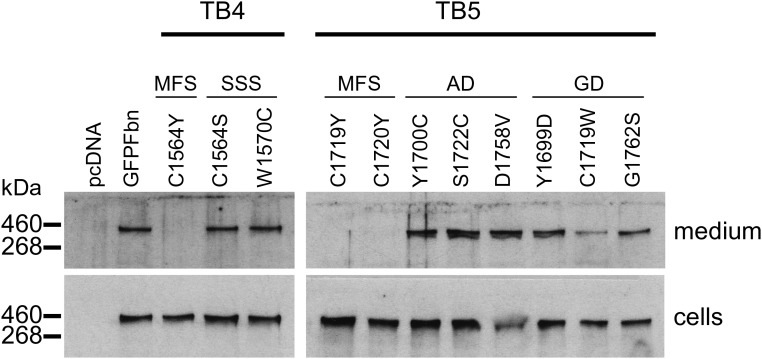
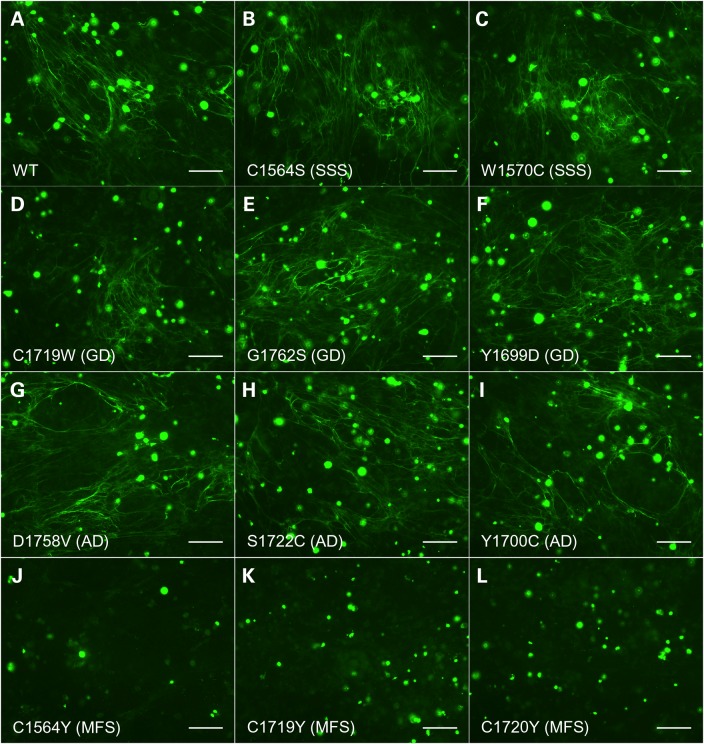
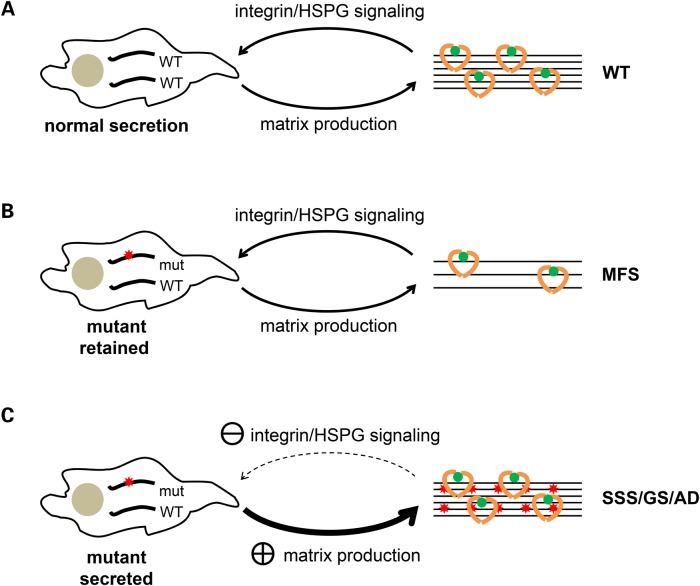
Fibrillin-1 is the major component of the 10-12 nm diameter extracellular matrix microfibrils. The majority of mutations affecting the human fibrillin-1 gene, FBN1, result in Marfan syndrome (MFS), a common connective tissue disorder characterised by tall stature, ocular and cardiovascular defects. Recently, stiff skin syndrome (SSS) and a group of syndromes known collectively as the acromelic dysplasias, which typically result in short stature, skin thickening and joint stiffness, have been linked to FBN1 mutations that affect specific domains of the fibrillin-1 protein. Despite their apparent phenotypic differences, dysregulation of transforming growth factor β (TGFβ) is a common factor in all of these disorders. Using a newly developed assay to track the secretion and incorporation of full-length, GFP-tagged fibrillin-1 into the extracellular matrix, we investigated whether or not there were differences in the secretion and microfibril assembly profiles of fibrillin-1 variants containing substitutions associated with MFS, SSS or the acromelic dysplasias. We show that substitutions in fibrillin-1 domains TB4 and TB5 that cause SSS and the acromelic dysplasias do not prevent fibrillin-1 from being secreted or assembled into microfibrils, whereas MFS-associated substitutions in these domains result in a loss of recombinant protein in the culture medium and no association with microfibrils. These results suggest fundamental differences in the dominant pathogenic mechanisms underlying MFS, SSS and the acromelic dysplasias, which give rise to TGFβ dysregulation associated with these diseases.
© The Author 2015. Published by Oxford University Press.
Figures


Similar articles
-
New insights into the structure, assembly and biological roles of 10-12 nm connective tissue microfibrils from fibrillin-1 studies.Biochem J. 2016 Apr 1;473(7):827-38. doi: 10.1042/BJ20151108. Biochem J. 2016. PMID: 27026396 Review.
-
Fibrillin protein pleiotropy: Acromelic dysplasias.Matrix Biol. 2019 Jul;80:6-13. doi: 10.1016/j.matbio.2018.09.005. Epub 2018 Sep 13. Matrix Biol. 2019. PMID: 30219651 Review.
-
Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias.Am J Hum Genet. 2011 Jul 15;89(1):7-14. doi: 10.1016/j.ajhg.2011.05.012. Epub 2011 Jun 16. Am J Hum Genet. 2011. PMID: 21683322 Free PMC article.
-
The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation?Matrix Biol. 2015 Sep;47:3-12. doi: 10.1016/j.matbio.2015.05.002. Epub 2015 May 7. Matrix Biol. 2015. PMID: 25957947 Review.
-
Microenvironmental regulation by fibrillin-1.PLoS Genet. 2012 Jan;8(1):e1002425. doi: 10.1371/journal.pgen.1002425. Epub 2012 Jan 5. PLoS Genet. 2012. PMID: 22242013 Free PMC article.
Cited by
-
Genotype-phenotype correlations of marfan syndrome and related fibrillinopathies: Phenomenon and molecular relevance.Front Genet. 2022 Aug 16;13:943083. doi: 10.3389/fgene.2022.943083. eCollection 2022. Front Genet. 2022. PMID: 36176293 Free PMC article. Review.
-
Middle-Aged Female Diagnosed With Widespread Stiff Skin Syndrome.Arch Rheumatol. 2018 Mar 23;33(4):491-493. doi: 10.5606/ArchRheumatol.2018.6805. eCollection 2018 Dec. Arch Rheumatol. 2018. PMID: 30874234 Free PMC article. No abstract available.
-
Cooperative Mechanism of ADAMTS/ ADAMTSL and Fibrillin-1 in the Marfan Syndrome and Acromelic Dysplasias.Front Genet. 2021 Nov 29;12:734718. doi: 10.3389/fgene.2021.734718. eCollection 2021. Front Genet. 2021. PMID: 34912367 Free PMC article. Review.
-
POGLUT2 and POGLUT3 O-glucosylate multiple EGF repeats in fibrillin-1, -2, and LTBP1 and promote secretion of fibrillin-1.J Biol Chem. 2021 Sep;297(3):101055. doi: 10.1016/j.jbc.2021.101055. Epub 2021 Aug 17. J Biol Chem. 2021. PMID: 34411563 Free PMC article.
-
Fibrillin microfibrils in bone physiology.Matrix Biol. 2016 May-Jul;52-54:191-197. doi: 10.1016/j.matbio.2015.09.004. Epub 2015 Sep 25. Matrix Biol. 2016. PMID: 26408953 Free PMC article. Review.
References
-
- Jones C.J., Sear C.H., Grant M.E. (1980) An ultrastructural study of fibroblasts derived from bovine ligamentum nuchae and their capacity for elastogenesis in culture. J. Pathol., 131, 35–53. - PubMed
-
- Kewley M.A., Williams G., Steven F.S. (1978) Studies of elastic tissue formation in the developing bovine ligamentum nuchae. J. Pathol., 124, 95–101. - PubMed
-
- Bax D.V., Bernard S.E., Lomas A., Morgan A., Humphries J., Shuttleworth C.A., Humphries M.J., Kielty C.M. (2003) Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by alpha 5 beta 1 and alpha v beta 3 integrins. J. Biol. Chem., 278, 34605–34616. - PubMed
-
- Jovanovic J., Takagi J., Choulier L., Abrescia N.G., Stuart D.I., van der Merwe P.A., Mardon H.J., Handford P.A. (2007) alphaVbeta6 is a novel receptor for human fibrillin-1. Comparative studies of molecular determinants underlying integrin-rgd affinity and specificity. J. Biol. Chem., 282, 6743–6751. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
