

The Autoimmunity-Associated Gene CLEC16A Modulates Thymic Epithelial Cell Autophagy and Alters T Cell Selection
- PMID: 25979422
- PMCID: PMC4439257
- DOI: 10.1016/j.immuni.2015.04.011
The Autoimmunity-Associated Gene CLEC16A Modulates Thymic Epithelial Cell Autophagy and Alters T Cell Selection
Abstract
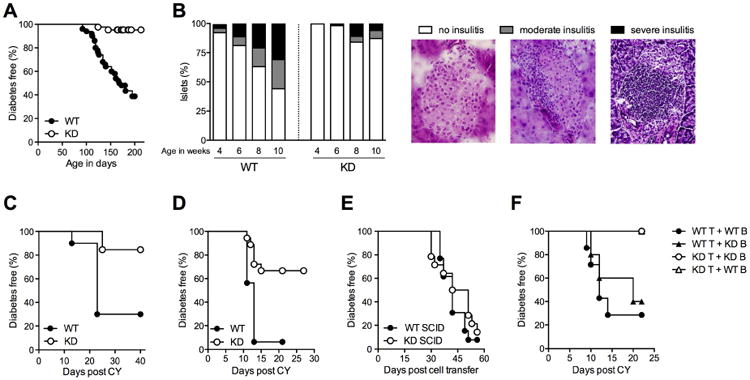
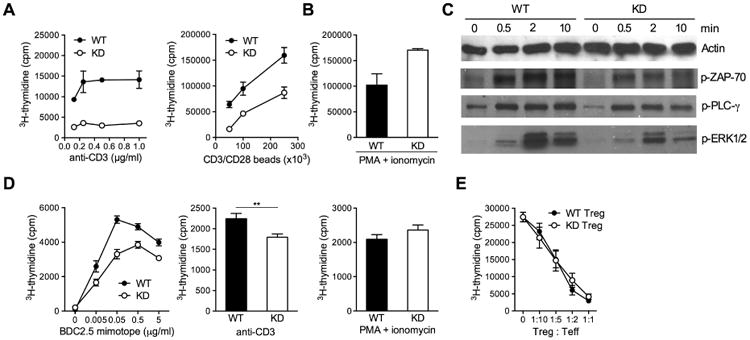
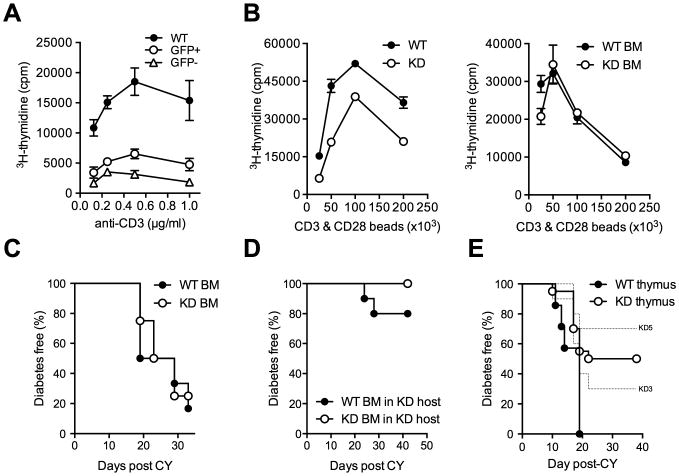
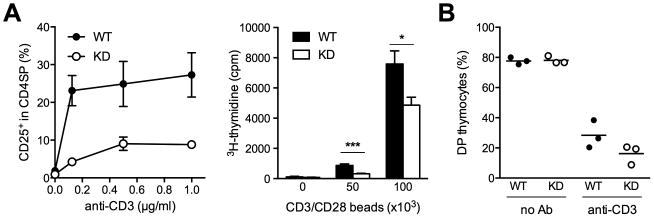
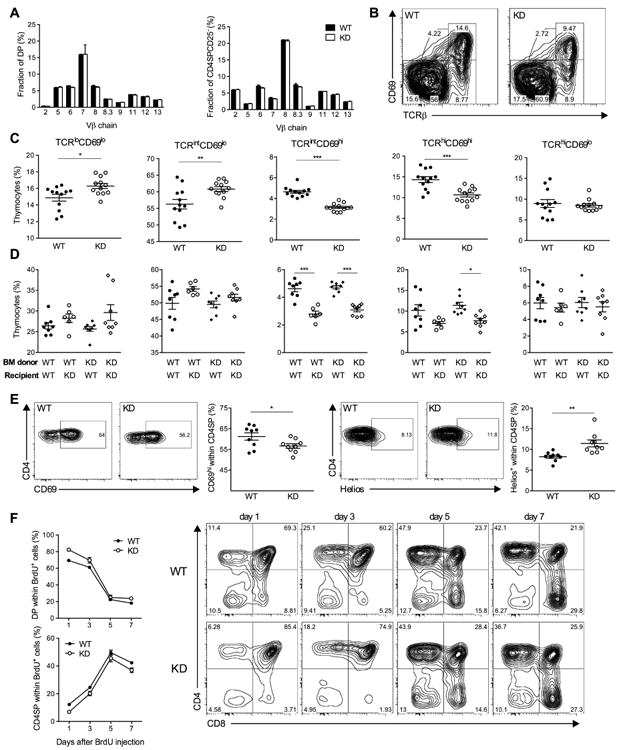
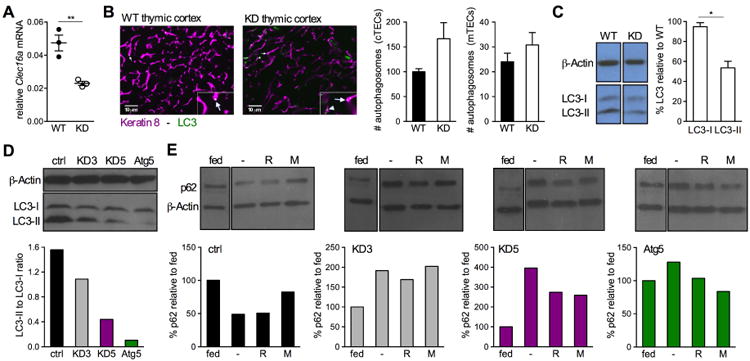
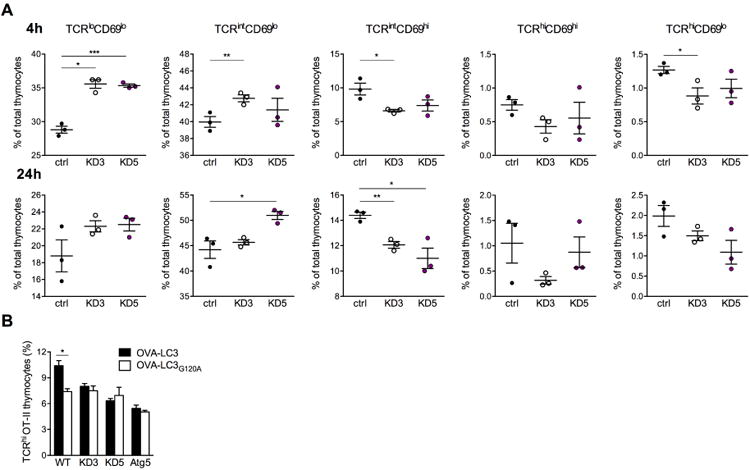
CLEC16A variation has been associated with multiple immune-mediated diseases, including type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, celiac disease, Crohn's disease, Addison's disease, primary biliary cirrhosis, rheumatoid arthritis, juvenile idiopathic arthritis, and alopecia areata. Despite strong genetic evidence implicating CLEC16A in autoimmunity, this gene's broad association with disease remains unexplained. We generated Clec16a knock-down (KD) mice in the nonobese diabetic (NOD) model for type 1 diabetes and found that Clec16a silencing protected against autoimmunity. Disease protection was attributable to T cell hyporeactivity, which was secondary to changes in thymic epithelial cell (TEC) stimuli that drive thymocyte selection. Our data indicate that T cell selection and reactivity were impacted by Clec16a variation in thymic epithelium owing to Clec16a's role in TEC autophagy. These findings provide a functional link between human CLEC16A variation and the immune dysregulation that underlies the risk of autoimmunity.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
References
-
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. - PubMed
-
- Brazil MI, Weiss S, Stockinger B. Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol. 1997;27:1506–1514. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
