

Nuclear EGFR impairs ASPP2-p53 complex-induced apoptosis by inducing SOS1 expression in hepatocellular carcinoma
- PMID: 25980493
- PMCID: PMC4599285
- DOI: 10.18632/oncotarget.3757
Nuclear EGFR impairs ASPP2-p53 complex-induced apoptosis by inducing SOS1 expression in hepatocellular carcinoma
Abstract
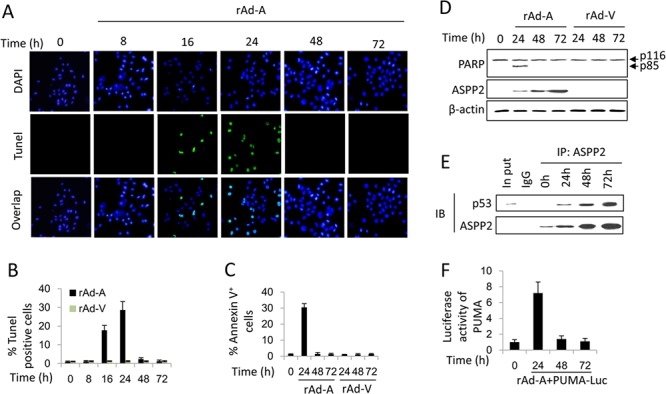
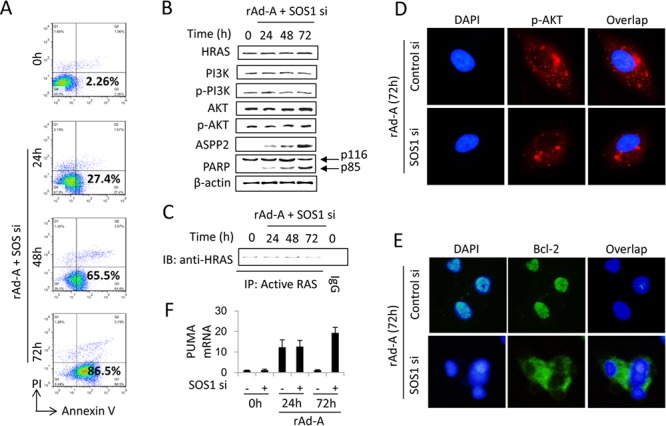
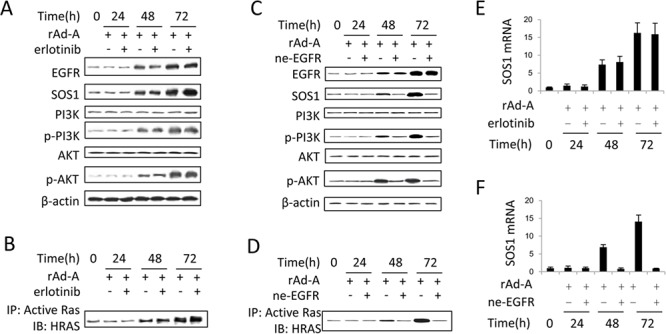
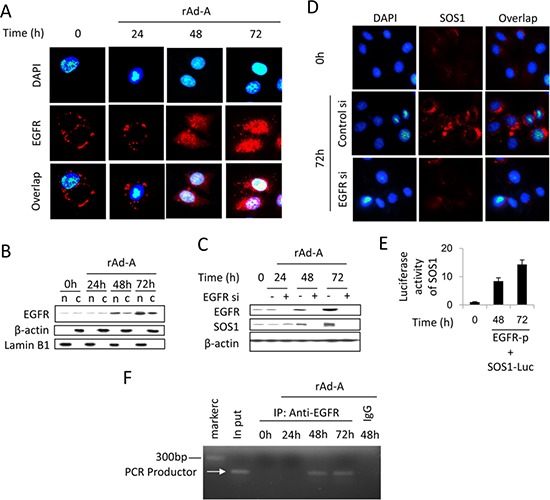
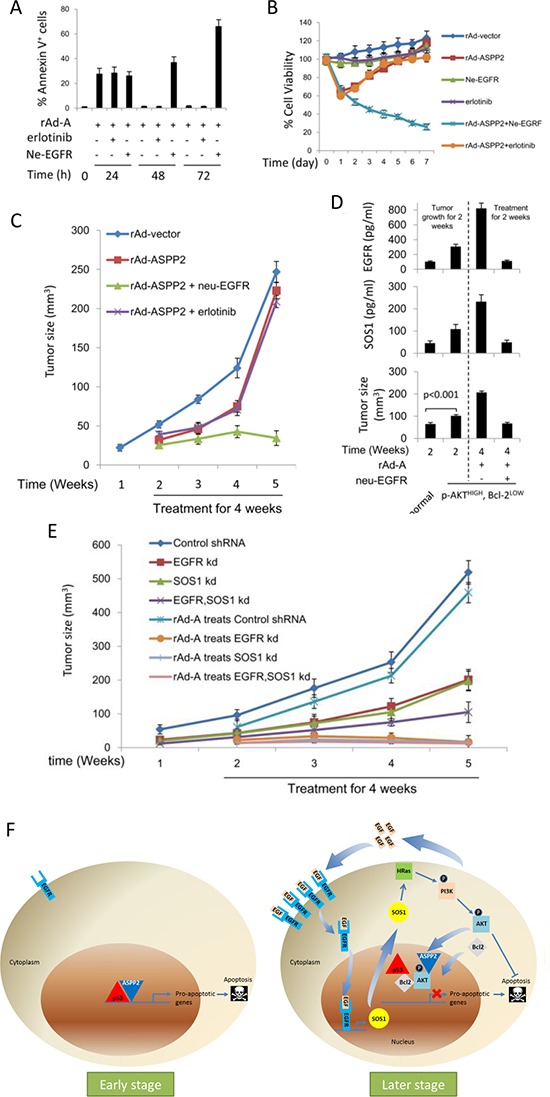
ASPP2 can bind to p53 and enhance the apoptotic capabilities of p53 by guiding it to the promoters of pro-apoptotic genes. Here, ASPP2 overexpression for 24 hours transiently induced apoptosis in hepatoma cells by enhancing the transactivation of p53 on pro-apoptotic gene promoters. However, long-term ASPP2 overexpression (more than 48 hours) failed to induce apoptosis because p53 was released from the pro-apoptotic gene promoters. In non-apoptotic cells, nuclear EGFR induced SOS1 expression by directly binding to the SOS1 promoter. SOS1 activated the HRAS/PI3K/AKT pathway and resulted in nuclear translocation of p-AKT and Bcl-2. The interaction between p-AKT and ASPP2 facilitates Bcl-2 binding to p53, which releases p53 from the pro-apoptotic gene promoters. The in vivo assay demonstrated that EGFR/SOS1-promoted growth of nuclear p-AKT+, Bcl-2+ cells results in the resistance of hepatoma cells to ASPP2-p53 complex-induced apoptosis and that blocking nuclear translocation of EGFR dramatically improves and enhances the pro-apoptotic function of ASPP2. Finally, the activation of the HRAS/PI3K/AKT pathway by EGFR-induced SOS1 also inhibits cisplatin-induced apoptosis, suggesting a common apoptosis-evasion mechanism in hepatoma cells. Because evasion of apoptosis contributes to treatment resistance in hepatoma, our results also support further investigation of combined therapeutic blockade of EGFR and SOS1.
Keywords: ASPP2; EGFR; SOS1; apoptosis; p53.
Conflict of interest statement
All authors declare no conflict of interest.
Figures


Similar articles
-
Amphiregulin impairs apoptosis-stimulating protein 2 of p53 overexpression-induced apoptosis in hepatoma cells.Tumour Biol. 2017 Mar;39(3):1010428317695026. doi: 10.1177/1010428317695026. Tumour Biol. 2017. PMID: 28351301
-
ASPP2 enhances chemotherapeutic sensitivity through the down-regulation of XIAP expression in a p53 independent manner in hepatocellular carcinoma.Biochem Biophys Res Commun. 2019 Jan 15;508(3):769-774. doi: 10.1016/j.bbrc.2018.11.181. Epub 2018 Dec 6. Biochem Biophys Res Commun. 2019. PMID: 30528232
-
CHOP mediates ASPP2-induced autophagic apoptosis in hepatoma cells by releasing Beclin-1 from Bcl-2 and inducing nuclear translocation of Bcl-2.Cell Death Dis. 2014 Jul 17;5(7):e1323. doi: 10.1038/cddis.2014.276. Cell Death Dis. 2014. PMID: 25032846 Free PMC article.
-
New insights into the expanding complexity of the tumor suppressor ASPP2.Cell Cycle. 2009 Sep 15;8(18):2871-6. doi: 10.4161/cc.8.18.9474. Epub 2009 Sep 8. Cell Cycle. 2009. PMID: 19657229 Review.
-
ASPP2: a gene that controls life and death in vivo.Cell Cycle. 2006 Oct;5(19):2187-90. doi: 10.4161/cc.5.19.3266. Epub 2006 Oct 1. Cell Cycle. 2006. PMID: 16969108 Review.
Cited by
-
TP53BP2: Roles in suppressing tumorigenesis and therapeutic opportunities.Genes Dis. 2022 Sep 5;10(5):1982-1993. doi: 10.1016/j.gendis.2022.08.014. eCollection 2023 Sep. Genes Dis. 2022. PMID: 37492707 Free PMC article. Review.
-
Nuclear Epidermal Growth Factor Receptor Overexpression as a Survival Predictor in Oral Squamous Cell Carcinoma.Int J Mol Sci. 2023 Mar 18;24(6):5816. doi: 10.3390/ijms24065816. Int J Mol Sci. 2023. PMID: 36982894 Free PMC article.
-
Runs of homozygosity associate with decreased risks of lung cancer in never-smoking East Asian females.J Cancer. 2018 Oct 5;9(21):3858-3866. doi: 10.7150/jca.22855. eCollection 2018. J Cancer. 2018. PMID: 30410588 Free PMC article.
-
Relationship between EGFR expression and subcellular localization with cancer development and clinical outcome.Oncotarget. 2019 Mar 8;10(20):1918-1931. doi: 10.18632/oncotarget.26727. eCollection 2019 Mar 8. Oncotarget. 2019. PMID: 30956774 Free PMC article.
-
DNA damage regulated autophagy modulator 1 recovers the function of apoptosis-stimulating of p53 protein 2 on inducing apoptotic cell death in Huh7.5 cells.Oncol Lett. 2018 Jun;15(6):9333-9338. doi: 10.3892/ol.2018.8453. Epub 2018 Apr 10. Oncol Lett. 2018. PMID: 29844830 Free PMC article.
References
-
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. - PubMed
-
- Fulda S. Tumor resistance to apoptosis. Int J Cancer. 2009;124:511–515. - PubMed
-
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. - PubMed
-
- Penuelas I, Mazzolini G, Boan JF, Sangro B, Marti-Climent J, Ruiz M, et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology. 2005;128:1787–1795. - PubMed
-
- Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33:162–167. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
