

De novo point mutations in patients diagnosed with ataxic cerebral palsy
- PMID: 25981959
- PMCID: PMC4572487
- DOI: 10.1093/brain/awv117
De novo point mutations in patients diagnosed with ataxic cerebral palsy
Erratum in
-
Corrigendum.Brain. 2016 Feb;139(Pt 2):e14. doi: 10.1093/brain/awv309. Brain. 2016. PMID: 26912521 Free PMC article. No abstract available.
Abstract
Cerebral palsy is a sporadic disorder with multiple likely aetiologies, but frequently considered to be caused by birth asphyxia. Genetic investigations are rarely performed in patients with cerebral palsy and there is little proven evidence of genetic causes. As part of a large project investigating children with ataxia, we identified four patients in our cohort with a diagnosis of ataxic cerebral palsy. They were investigated using either targeted next generation sequencing or trio-based exome sequencing and were found to have mutations in three different genes, KCNC3, ITPR1 and SPTBN2. All the mutations were de novo and associated with increased paternal age. The mutations were shown to be pathogenic using a combination of bioinformatics analysis and in vitro model systems. This work is the first to report that the ataxic subtype of cerebral palsy can be caused by de novo dominant point mutations, which explains the sporadic nature of these cases. We conclude that at least some subtypes of cerebral palsy may be caused by de novo genetic mutations and patients with a clinical diagnosis of cerebral palsy should be genetically investigated before causation is ascribed to perinatal asphyxia or other aetiologies.
Keywords: ataxia; cerebral palsy; de novo; intellectual disability.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
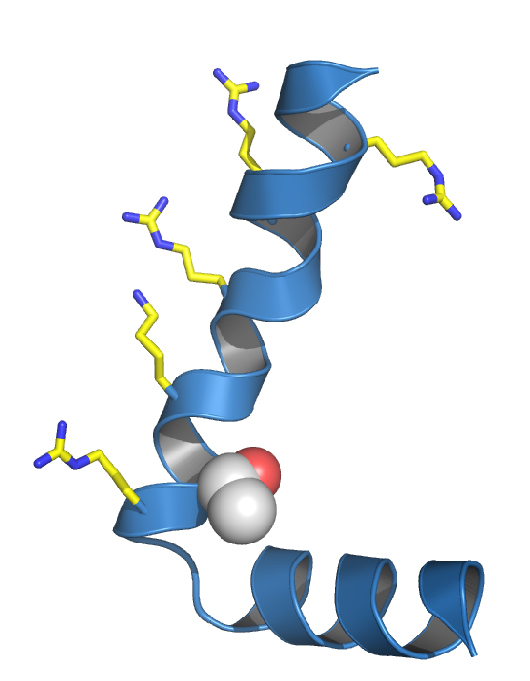
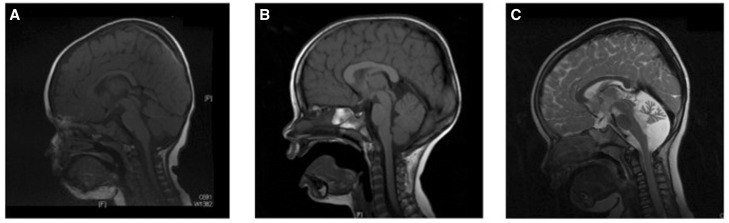
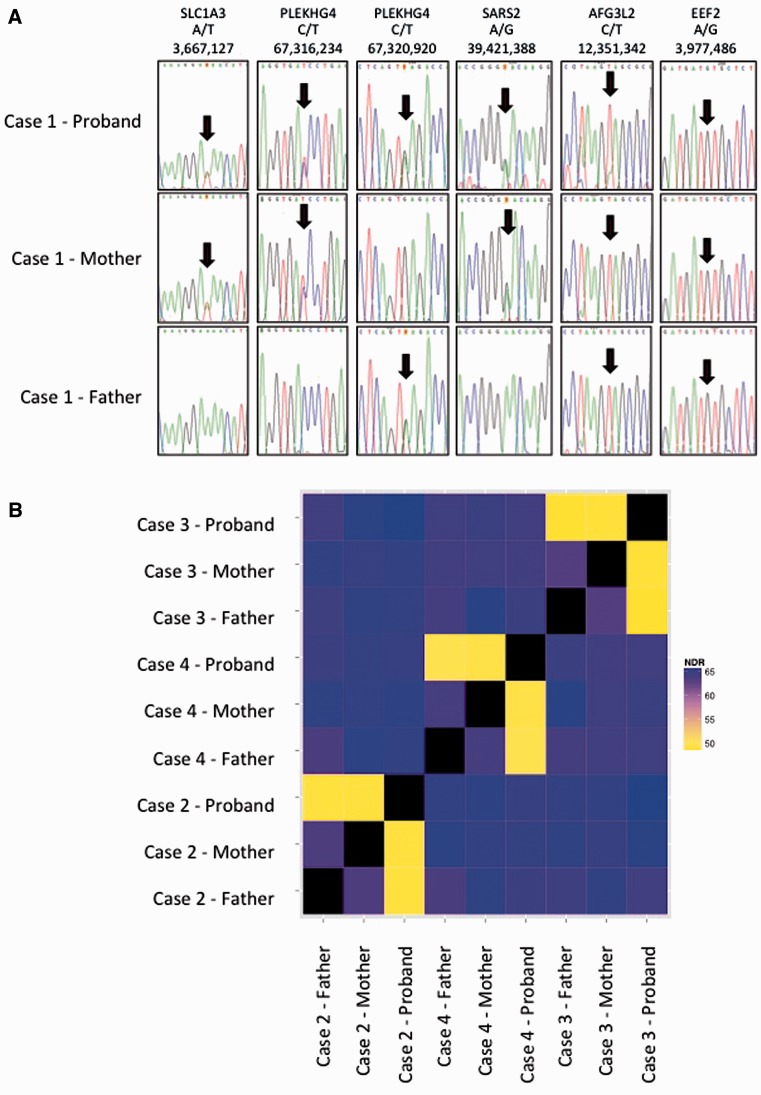
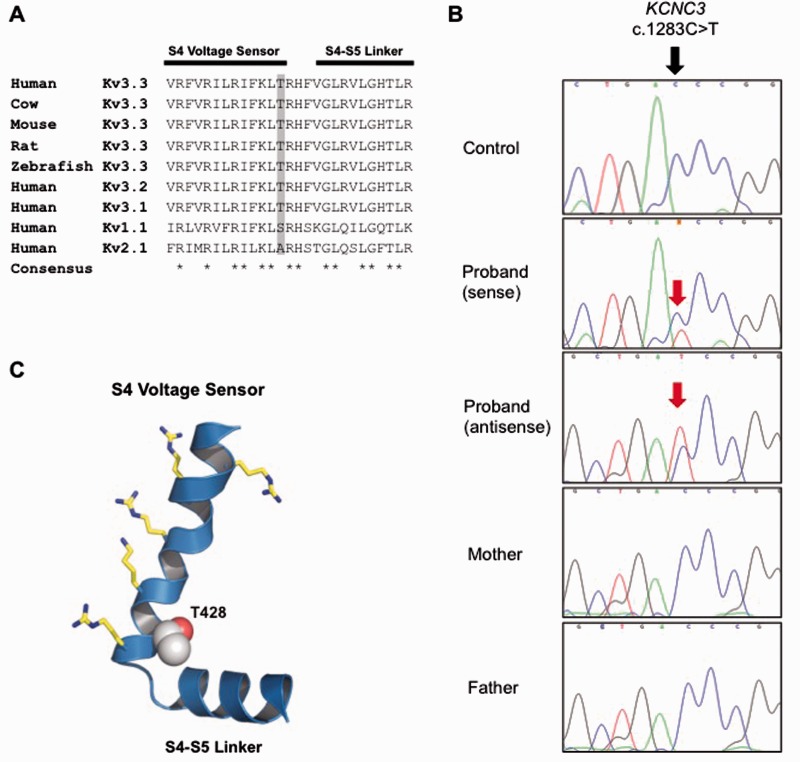
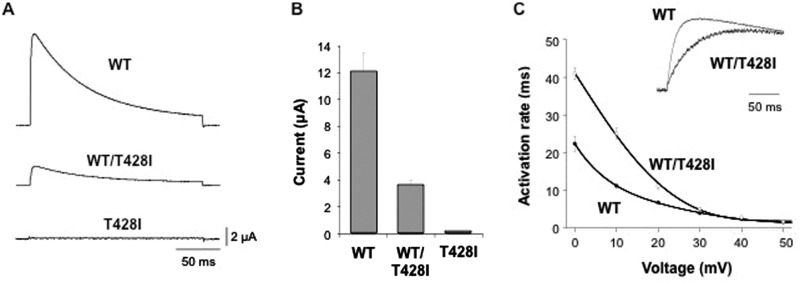
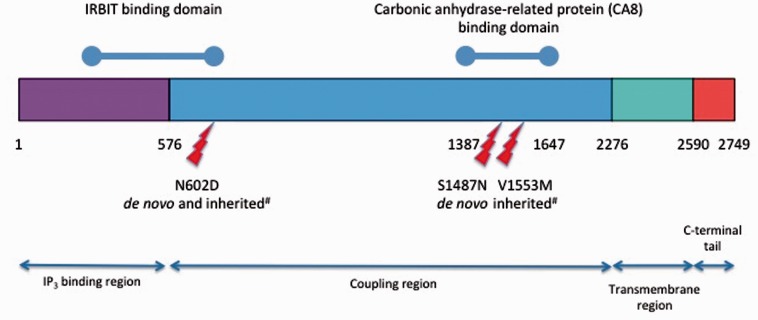
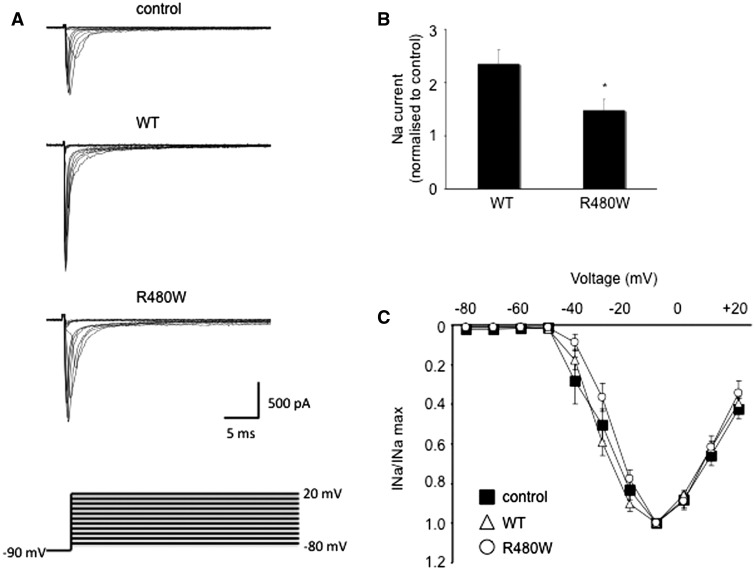
References
-
- Ando H, Mizutani A, Kiefer H, Tsuzurugi D, Michikawa T, Mikoshiba K. IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Mol Cell 2006; 22: 795–806. - PubMed
-
- Bauer P, Schols L, Riess O. Spectrin mutations in spinocerebellar ataxia (SCA). Bioessays 2006; 28: 785–7. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
