

Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection
- PMID: 25984700
- PMCID: PMC4856034
- DOI: 10.1038/nmeth.3407
Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection
Abstract
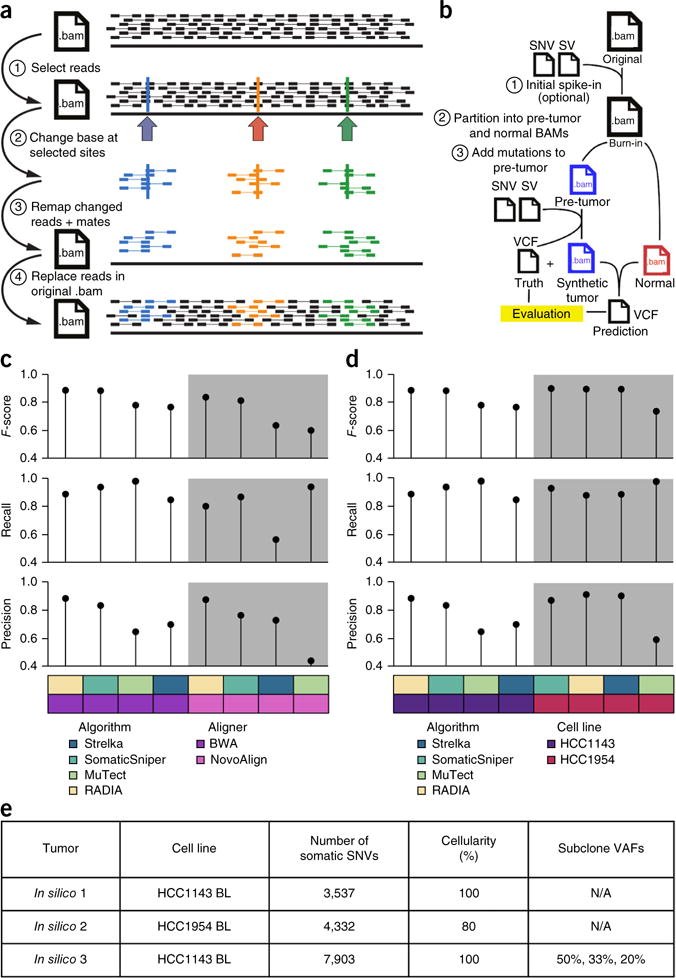
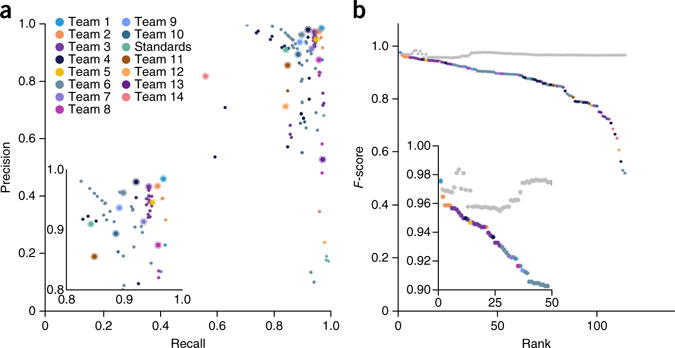
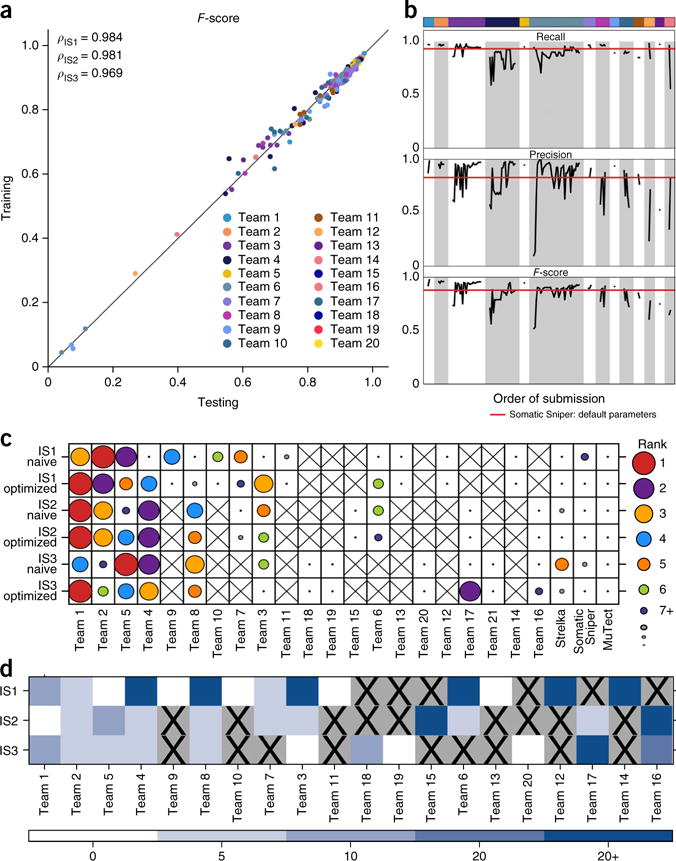
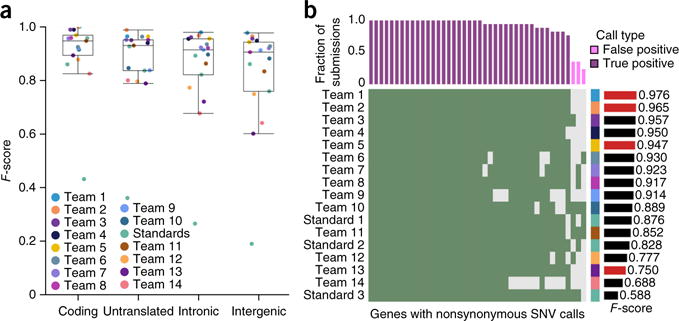
The detection of somatic mutations from cancer genome sequences is key to understanding the genetic basis of disease progression, patient survival and response to therapy. Benchmarking is needed for tool assessment and improvement but is complicated by a lack of gold standards, by extensive resource requirements and by difficulties in sharing personal genomic information. To resolve these issues, we launched the ICGC-TCGA DREAM Somatic Mutation Calling Challenge, a crowdsourced benchmark of somatic mutation detection algorithms. Here we report the BAMSurgeon tool for simulating cancer genomes and the results of 248 analyses of three in silico tumors created with it. Different algorithms exhibit characteristic error profiles, and, intriguingly, false positives show a trinucleotide profile very similar to one found in human tumors. Although the three simulated tumors differ in sequence contamination (deviation from normal cell sequence) and in subclonality, an ensemble of pipelines outperforms the best individual pipeline in all cases. BAMSurgeon is available at https://github.com/adamewing/bamsurgeon/.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
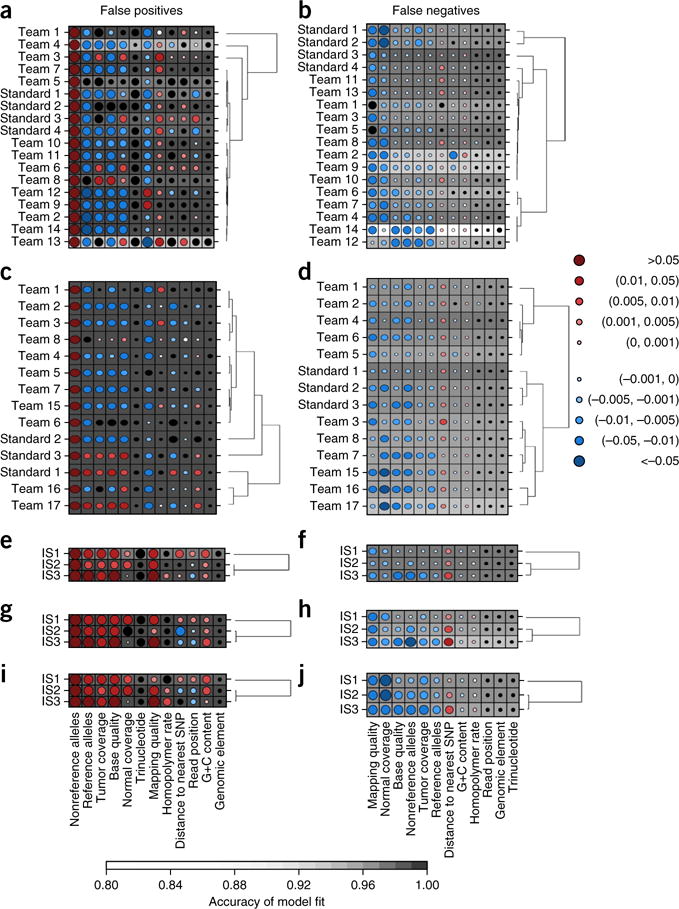
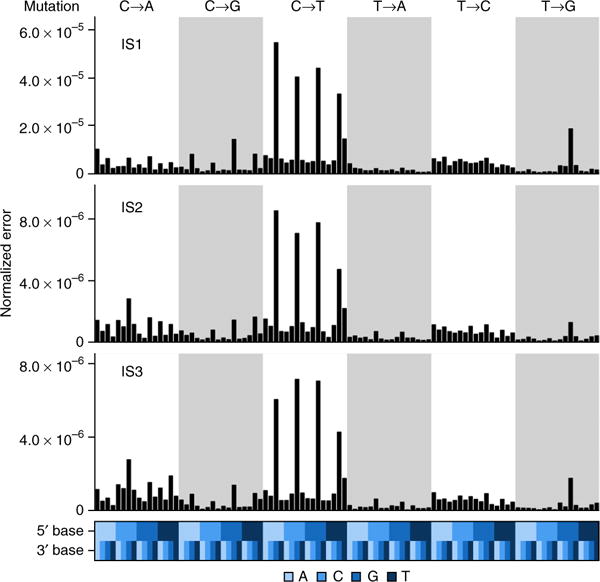
References
-
- Anonymous. Adaptive BATTLE trial uses biomarkers to guide lung cancer treatment. Nat Rev Drug Discov. 2010;9:423. - PubMed
-
- Tran B, et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer. 2013;132:1547–1555. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
