

The GST-BHMT assay reveals a distinct mechanism underlying proteasome inhibition-induced macroautophagy in mammalian cells
- PMID: 25984893
- PMCID: PMC4509454
- DOI: 10.1080/15548627.2015.1034402
The GST-BHMT assay reveals a distinct mechanism underlying proteasome inhibition-induced macroautophagy in mammalian cells
Abstract
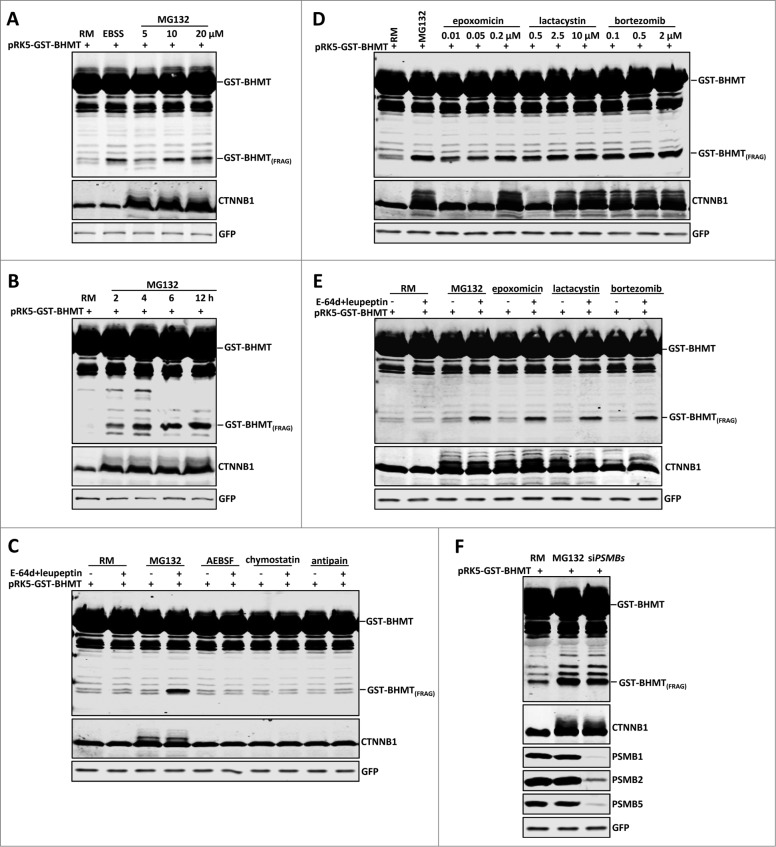
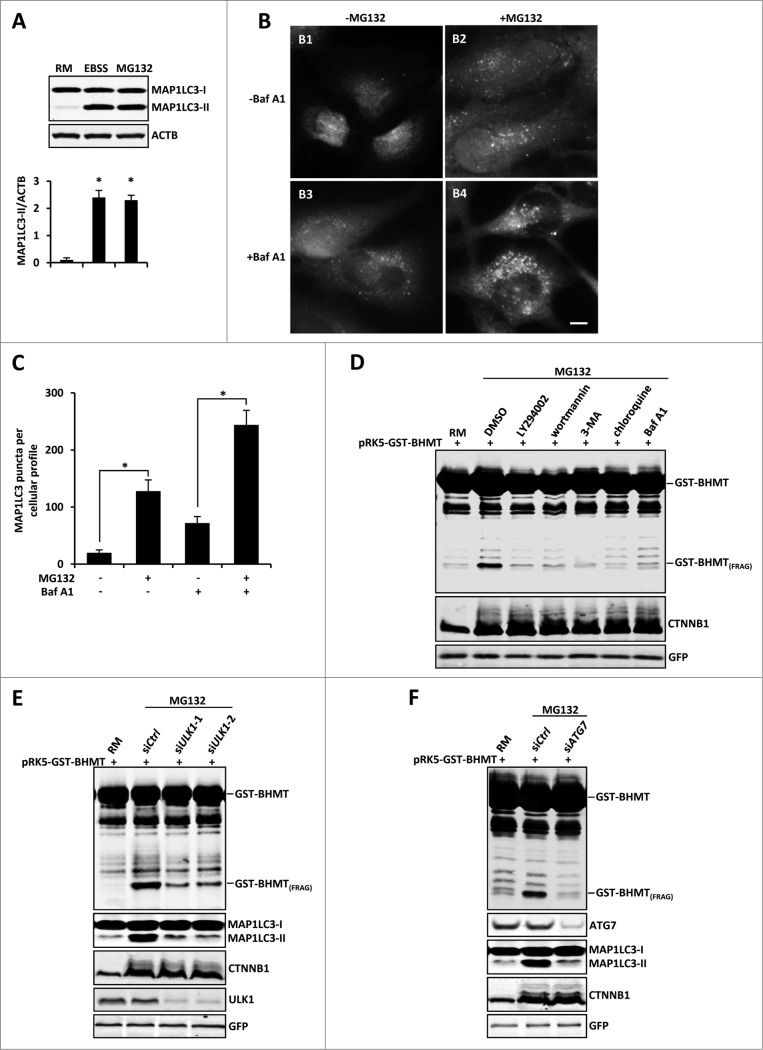
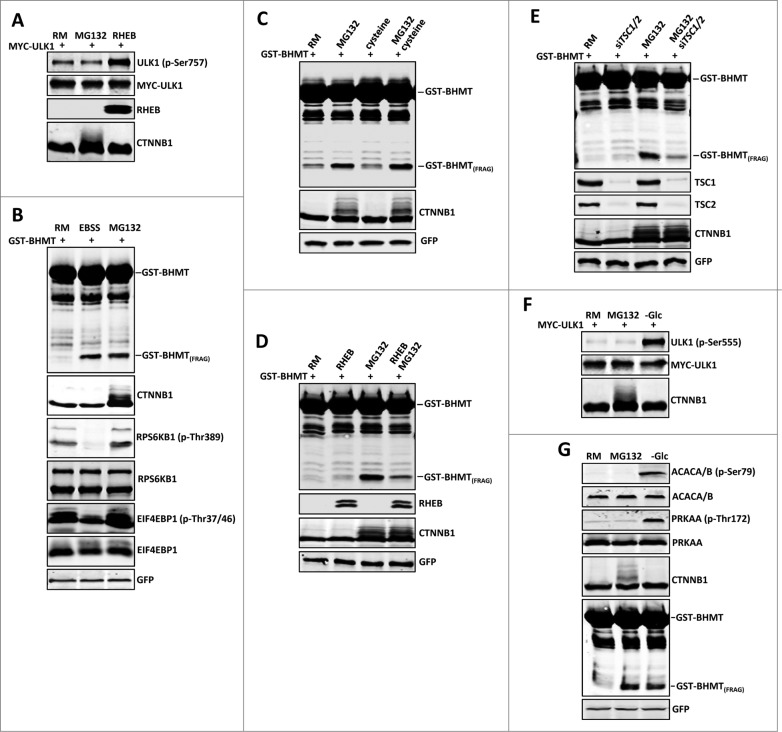
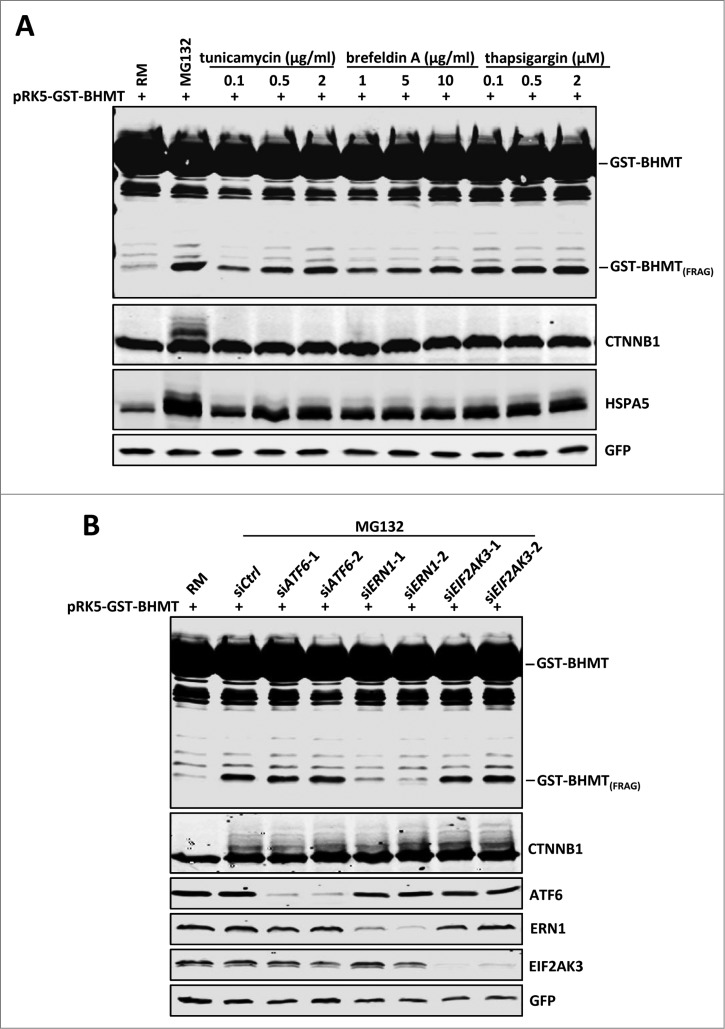
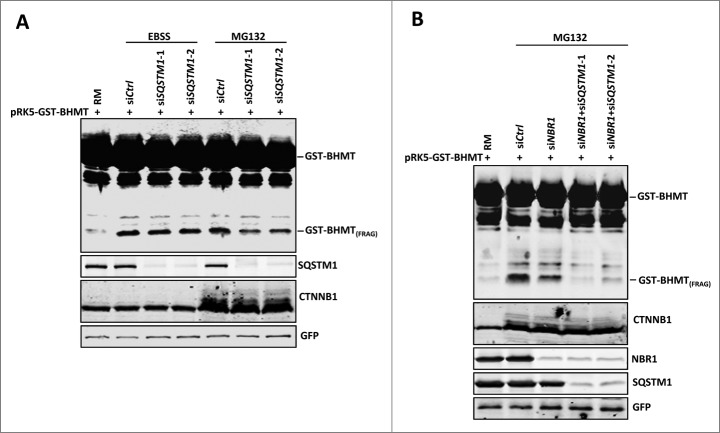
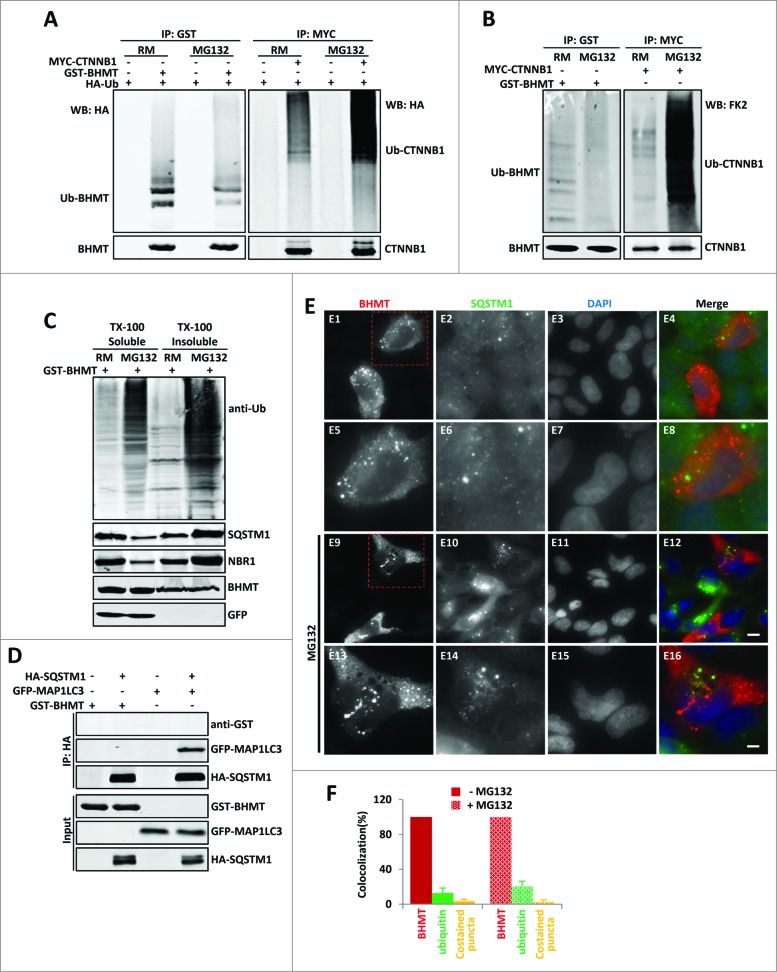
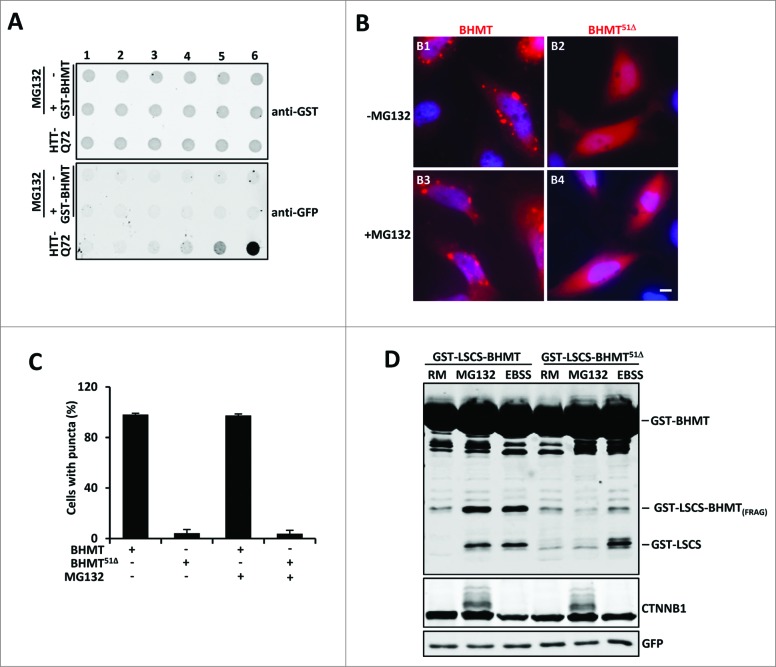
By monitoring the fragmentation of a GST-BHMT (a protein fusion of glutathionine S-transferase N-terminal to betaine-homocysteine S-methyltransferase) reporter in lysosomes, the GST-BHMT assay has previously been established as an endpoint, cargo-based assay for starvation-induced autophagy that is largely nonselective. Here, we demonstrate that under nutrient-rich conditions, proteasome inhibition by either pharmaceutical or genetic manipulations induces similar autophagy-dependent GST-BHMT processing. However, mechanistically this proteasome inhibition-induced autophagy is different from that induced by starvation as it does not rely on regulation by MTOR (mechanistic target of rapamycin [serine/threonine kinase]) and PRKAA/AMPK (protein kinase, AMP-activated, α catalytic subunit), the upstream central sensors of cellular nutrition and energy status, but requires the presence of the cargo receptors SQSTM1/p62 (sequestosome 1) and NBR1 (neighbor of BRCA1 gene 1) that are normally involved in the selective autophagy pathway. Further, it depends on ER (endoplasmic reticulum) stress signaling, in particular ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1) and its main downstream effector MAPK8/JNK1 (mitogen-activated protein kinase 8), but not XBP1 (X-box binding protein 1), by regulating the phosphorylation-dependent disassociation of BCL2 (B-cell CLL/lymphoma 2) from BECN1 (Beclin 1, autophagy related). Moreover, the multimerization domain of GST-BHMT is required for its processing in response to proteasome inhibition, in contrast to its dispensable role in starvation-induced processing. Together, these findings support a model in which under nutrient-rich conditions, proteasome inactivation induces autophagy-dependent processing of the GST-BHMT reporter through a distinct mechanism that bears notable similarity with the yeast Cvt (cytoplasm-to-vacuole targeting) pathway, and suggest the GST-BHMT reporter might be employed as a convenient assay to study selective macroautophagy in mammalian cells.
Keywords: ACACA/B, acetyl-CoA carboxylase α/β; ACTB, actin, β; ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; ATG7, autophagy-related 7; BCL2, B-cell CLL/lymphoma 2; BECN1, Beclin 1, autophagy-related; BHMT; BHMT, betaine-homocysteine S-methyltransferase; Baf A1, bafilomycin A1; CTNNB1, catenin (cadherin-associated protein), β 1, 88kDa; Cvt, cytoplasm-to-vacuole-targeting; DDIT3, DNA-damage-inducible transcript 3; EBSS, Earle's Balanced Salt Solution; EIF2AK3, eukaryotic translation initiation factor 2-α, kinase 3; EIF4EBP1, eukaryotic translation initiation factor 4E binding protein 1; ER, endoplasmic reticulum; ERN1, endoplasmic reticulum to nucleus signaling 1; GST, glutathionine S-transferase; GST-BHMT(FRAG), an autophagy-mediated cleavage product of the GST-BHMT reporter; GST-BHMT, a fusion protein of glutathionine S-transferase N-terminal to betaine-homocysteine S-methyltransferase; HA, hemagglutinin; HSPA5, heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa); LSCS, linker-specific cleavage site; MAP1LC3, microtubule-associated protein 1 light chain 3; MAP2K7, mitogen-activated protein kinase kinase 7; MAPK8, mitogen-activated protein kinase 8; MTOR; MTOR, mechanistic target of rapamycin (serine/threonine kinase); MTORC1, MTOR complex 1; NBR1, neighbor of BRCA1 gene 1; P4HB, prolyl 4-hydroxylase, β polypeptide; PRKAA, protein kinase, AMP-activated, α catalytic subunit; PRKAA/AMPK; RHEB, Ras homolog enriched in brain; RM, rich medium; RPS6KB1, ribosomal protein S6 kinase, 70kDa, polypeptide 1; SQSTM1, sequestosome 1; TSC1/2, tuberous sclerosis 1/2; ULK1, unc-51 like autophagy activating kinase 1; UPR, unfolded protein response; UPS, ubiquitin proteasome system; XBP1, X-box binding protein 1; cargo receptors SQSTM1/p62 and NBR1; proteasome inhibition; selective macroautophagy.
Figures

Similar articles
-
MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint.Autophagy. 2020 Feb;16(2):271-288. doi: 10.1080/15548627.2019.1606647. Epub 2019 Apr 21. Autophagy. 2020. PMID: 31007149 Free PMC article.
-
RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy.Autophagy. 2015;11(7):975-94. doi: 10.1080/15548627.2015.1049800. Autophagy. 2015. PMID: 26018731 Free PMC article.
-
How autophagy controls the intestinal epithelial barrier.Autophagy. 2022 Jan;18(1):86-103. doi: 10.1080/15548627.2021.1909406. Epub 2021 Apr 27. Autophagy. 2022. PMID: 33906557 Free PMC article. Review.
-
DNA-dependent protein kinase regulates lysosomal AMP-dependent protein kinase activation and autophagy.Autophagy. 2020 Oct;16(10):1871-1888. doi: 10.1080/15548627.2019.1710430. Epub 2020 Jan 26. Autophagy. 2020. PMID: 31983282 Free PMC article.
-
Autophagy in the physiological endometrium and cancer.Autophagy. 2021 May;17(5):1077-1095. doi: 10.1080/15548627.2020.1752548. Epub 2020 May 13. Autophagy. 2021. PMID: 32401642 Free PMC article. Review.
Cited by
-
Proteostasis Dysfunction in Aged Mammalian Cells. The Stressful Role of Inflammation.Front Mol Biosci. 2021 Jun 17;8:658742. doi: 10.3389/fmolb.2021.658742. eCollection 2021. Front Mol Biosci. 2021. PMID: 34222330 Free PMC article. Review.
-
High wall shear stress-dependent podosome formation in a novel murine model of intracranial aneurysm.Front Stroke. 2024;3:1494559. doi: 10.3389/fstro.2024.1494559. Epub 2024 Nov 25. Front Stroke. 2024. PMID: 40236952 Free PMC article.
-
Dysregulation of autophagy during photoaging reduce oxidative stress and inflammatory damage caused by UV.Front Pharmacol. 2025 May 12;16:1562845. doi: 10.3389/fphar.2025.1562845. eCollection 2025. Front Pharmacol. 2025. PMID: 40421222 Free PMC article. Review.
-
Autophagy Induced by Proteasomal DUB Inhibitor NiPT Restricts NiPT-Mediated Cancer Cell Death.Front Oncol. 2020 Mar 27;10:348. doi: 10.3389/fonc.2020.00348. eCollection 2020. Front Oncol. 2020. PMID: 32292717 Free PMC article.
-
Identification of aberrantly methylated‑differentially expressed genes and gene ontology in prostate cancer.Mol Med Rep. 2020 Feb;21(2):744-758. doi: 10.3892/mmr.2019.10876. Epub 2019 Dec 11. Mol Med Rep. 2020. PMID: 31974616 Free PMC article.
References
-
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124–31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014 - DOI - PMC - PubMed
-
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X, et al.. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297–305; PMID:19258318; http://dx.doi.org/10.1074/jbc.M900573200 - DOI - PMC - PubMed
-
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67–93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910 - DOI - PMC - PubMed
-
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287–95; PMID:20083114; http://dx.doi.org/10.1016/j.febslet.2010.01.017 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous