

Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events
- PMID: 25985263
- PMCID: PMC4832928
- DOI: 10.1038/nbt.3242
Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events
Abstract
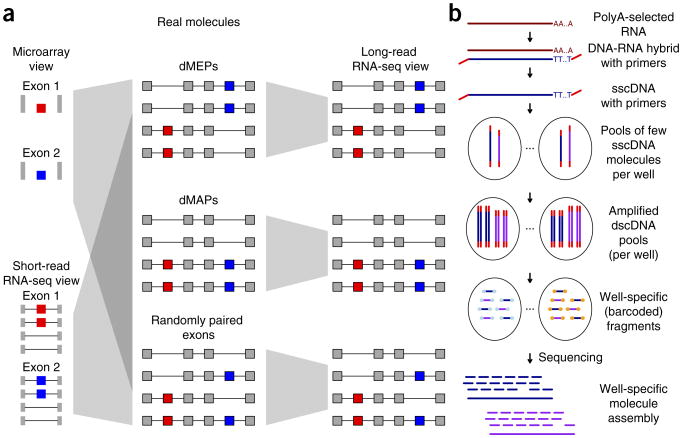
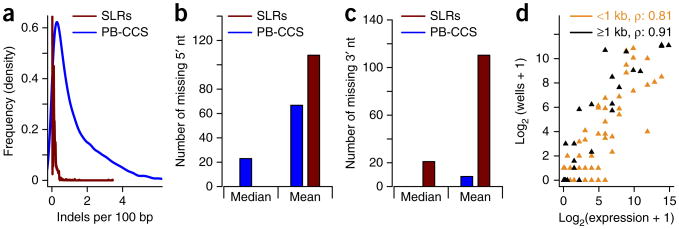
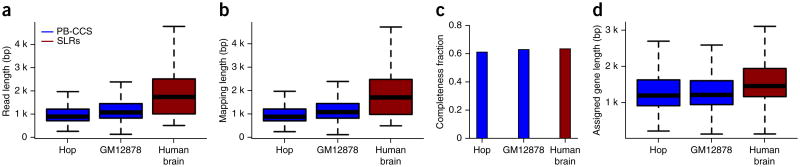
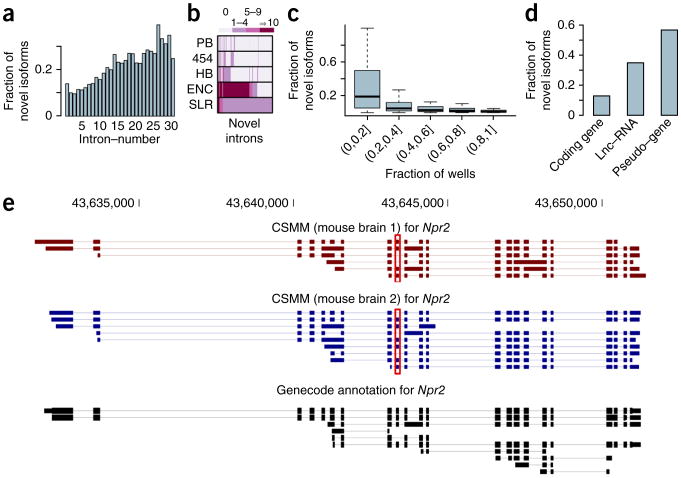
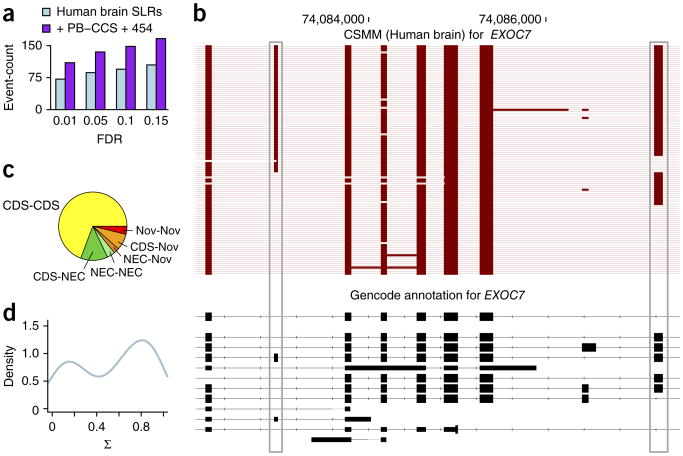
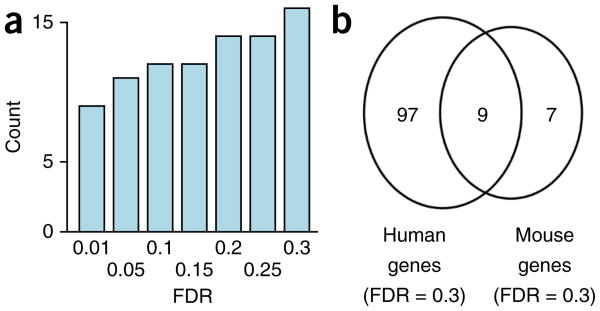
Alternative splicing shapes mammalian transcriptomes, with many RNA molecules undergoing multiple distant alternative splicing events. Comprehensive transcriptome analysis, including analysis of exon co-association in the same molecule, requires deep, long-read sequencing. Here we introduce an RNA sequencing method, synthetic long-read RNA sequencing (SLR-RNA-seq), in which small pools (≤1,000 molecules/pool, ≤1 molecule/gene for most genes) of full-length cDNAs are amplified, fragmented and short-read-sequenced. We demonstrate that these RNA sequences reconstructed from the short reads from each of the pools are mostly close to full length and contain few insertion and deletion errors. We report many previously undescribed isoforms (human brain: ∼13,800 affected genes, 14.5% of molecules; mouse brain ∼8,600 genes, 18% of molecules) and up to 165 human distant molecularly associated exon pairs (dMAPs) and distant molecularly and mutually exclusive pairs (dMEPs). Of 16 associated pairs detected in the mouse brain, 9 are conserved in human. Our results indicate conserved mechanisms that can produce distant but phased features on transcript and proteome isoforms.
Figures
References
-
- Kornblihtt AR, et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14:153–165. - PubMed
-
- Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2014;34:1–14. - PubMed
-
- Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. - PubMed
-
- Ben-Dov C, Hartmann B, Lundgren J, Valcárcel J. Genome-wide analysis of alternative pre-mRNA splicing. J Biol Chem. 2008;283:1229–1233. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
