

Allele-specific analysis of DNA replication origins in mammalian cells
- PMID: 25987481
- PMCID: PMC4479011
- DOI: 10.1038/ncomms8051
Allele-specific analysis of DNA replication origins in mammalian cells
Abstract
The mechanisms that control the location and timing of firing of replication origins are poorly understood. Using a novel functional genomic approach based on the analysis of SNPs and indels in phased human genomes, we observe that replication asynchrony is associated with small cumulative variations in the initiation efficiency of multiple origins between the chromosome homologues, rather than with the activation of dormant origins. Allele-specific measurements demonstrate that the presence of G-quadruplex-forming sequences does not correlate with the efficiency of initiation. Sequence analysis reveals that the origins are highly enriched in sequences with profoundly asymmetric G/C and A/T nucleotide distributions and are almost completely depleted of antiparallel triplex-forming sequences. We therefore propose that although G4-forming sequences are abundant in replication origins, an asymmetry in nucleotide distribution, which increases the propensity of origins to unwind and adopt non-B DNA structure, rather than the ability to form G4, is directly associated with origin activity.
Figures

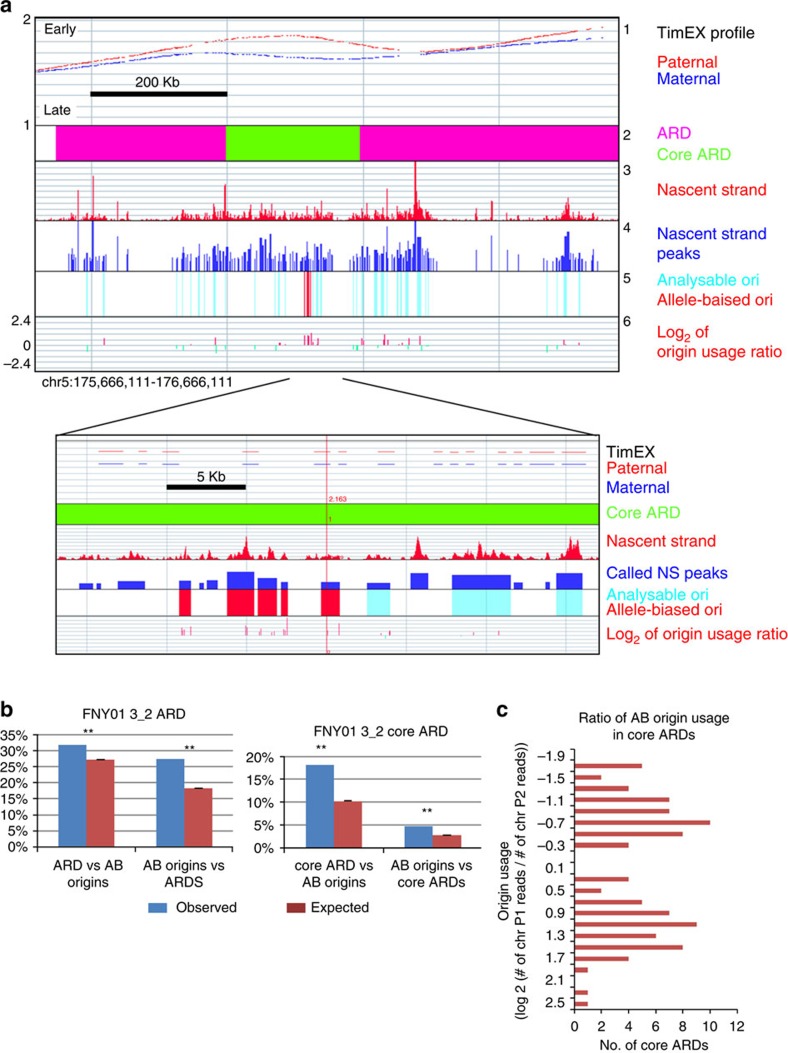


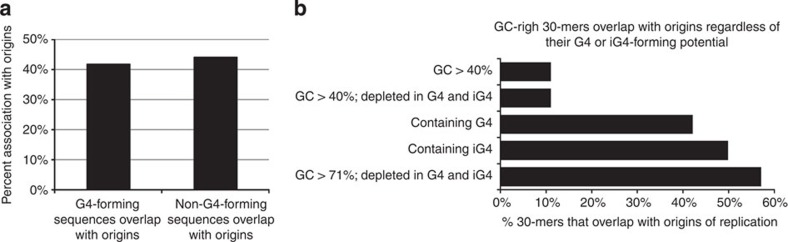

References
-
- Woodfine K. et al. Replication timing of the human genome. Hum. Mol. Genet. 13, 191–202 (2004). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
