

Targeted DNA degradation using a CRISPR device stably carried in the host genome
- PMID: 25988366
- PMCID: PMC4479009
- DOI: 10.1038/ncomms7989
Targeted DNA degradation using a CRISPR device stably carried in the host genome
Abstract
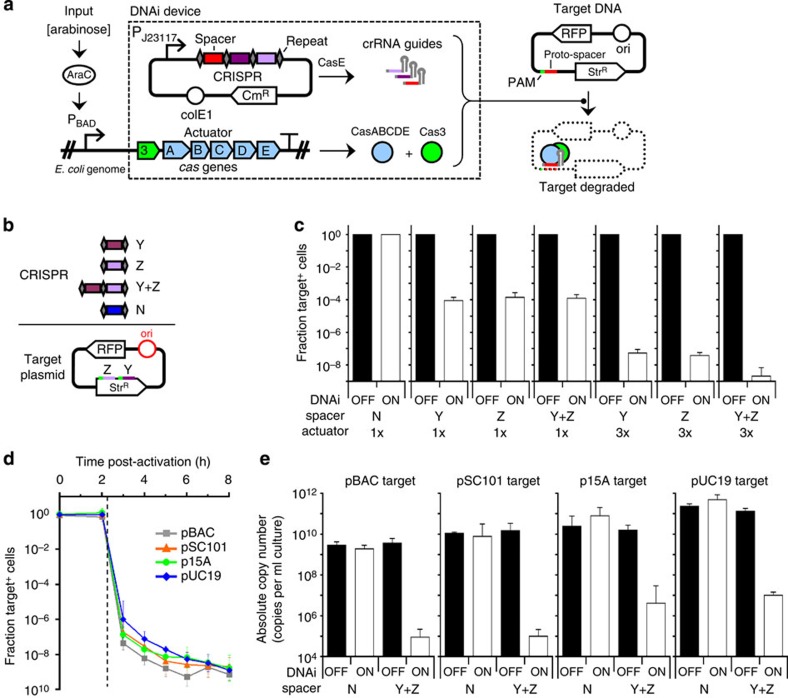
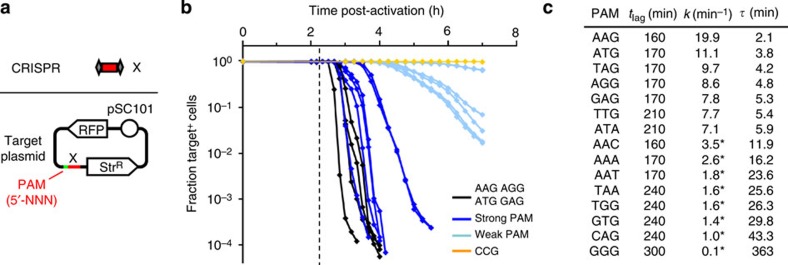
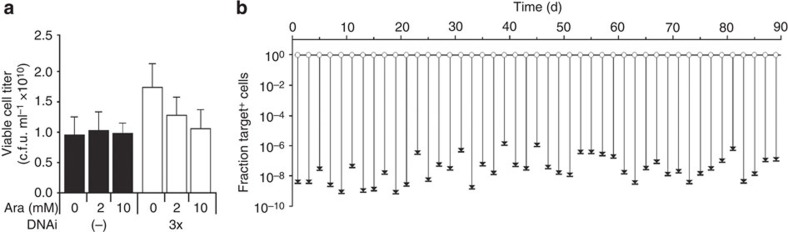
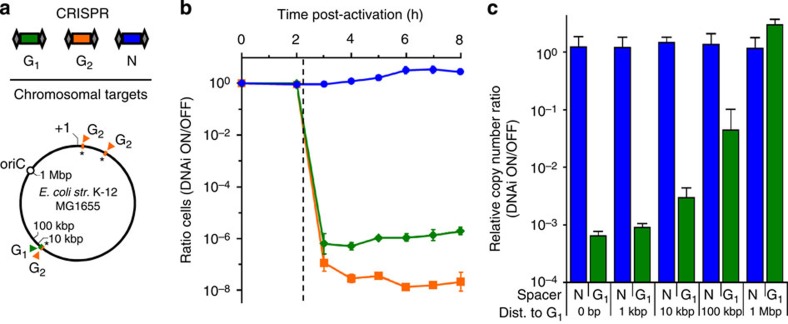
Once an engineered organism completes its task, it is useful to degrade the associated DNA to reduce environmental release and protect intellectual property. Here we present a genetically encoded device (DNAi) that responds to a transcriptional input and degrades user-defined DNA. This enables engineered regions to be obscured when the cell enters a new environment. DNAi is based on type-IE CRISPR biochemistry and a synthetic CRISPR array defines the DNA target(s). When the input is on, plasmid DNA is degraded 10(8)-fold. When the genome is targeted, this causes cell death, reducing viable cells by a factor of 10(8). Further, the CRISPR nuclease can direct degradation to specific genomic regions (for example, engineered or inserted DNA), which could be used to complicate recovery and sequencing efforts. DNAi can be stably carried in an engineered organism, with no impact on cell growth, plasmid stability or DNAi inducibility even after passaging for >2 months.
Figures
References
-
- Arkin A. Setting the standard in synthetic biology. Nat. Biotechnol. 26, 771–774 (2008). - PubMed
-
- Molin S. et al. Suicidal genetic elements and their use in biological containment of bacteria. Annu. Rev. Microbiol. 47, 139–166 (1993). - PubMed
-
- Isaacs F. J. et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 22, 841–847 (2004). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
