

Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy
- PMID: 25988613
- PMCID: PMC6272314
- DOI: 10.3390/molecules20058823
Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy
Abstract
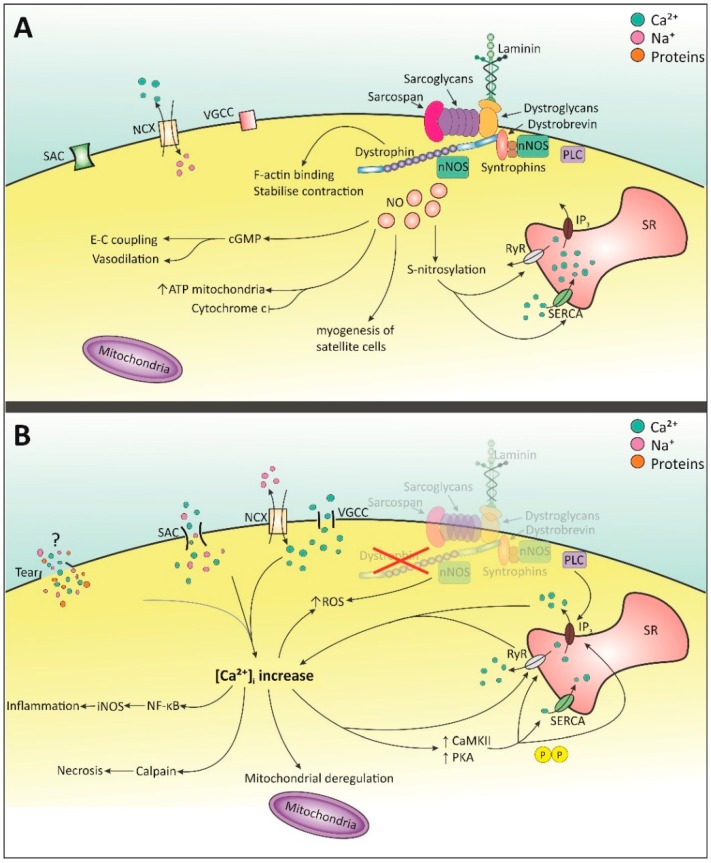
Duchenne muscular dystrophy (DMD) is a genetic muscle disorder caused by mutations in the Dmd gene resulting in the loss of the protein dystrophin. Patients do not only experience skeletal muscle degeneration, but also develop severe cardiomyopathy by their second decade, one of the main causes of death. The absence of dystrophin in the heart renders cardiomyocytes more sensitive to stretch-induced damage. Moreover, it pathologically alters intracellular calcium (Ca2+) concentration, neuronal nitric oxide synthase (nNOS) localization and mitochondrial function and leads to inflammation and necrosis, all contributing to the development of cardiomyopathy. Current therapies only treat symptoms and therefore the need for targeting the genetic defect is immense. Several preclinical therapies are undergoing development, including utrophin up-regulation, stop codon read-through therapy, viral gene therapy, cell-based therapy and exon skipping. Some of these therapies are undergoing clinical trials, but these have predominantly focused on skeletal muscle correction. However, improving skeletal muscle function without addressing cardiac aspects of the disease may aggravate cardiomyopathy and therefore it is essential that preclinical and clinical focus include improving heart function. This review consolidates what is known regarding molecular pathology of the DMD heart, specifically focusing on intracellular Ca2+, nNOS and mitochondrial dysregulation. It briefly discusses the current treatment options and then elaborates on the preclinical therapeutic approaches currently under development to restore dystrophin thereby improving pathology, with a focus on the heart.
Keywords: calcium; cell-based therapy; dystrophin; exon skipping; heart; mitochondria; nNOS; read-through; utrophin up-regulation; viral gene therapy.
Conflict of interest statement
MJAW has worked as a consultant for Sarepta Therapeutics.
Figures
References
-
- Mendell J.R., Province M.A., Moxley R.T., 3rd, Griggs R.C., Brooke M.H., Fenichel G.M., Miller J.P., Kaiser K.K., King W., Robison J., et al. Clinical investigation of Duchenne muscular dystrophy. A methodology for therapeutic trials based on natural history controls. Arch. Neurol. 1987;44:808–811. doi: 10.1001/archneur.1987.00520200012009. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
