

The impact of host genetic variation on infection with HIV-1
- PMID: 25988890
- PMCID: PMC6296468
- DOI: 10.1038/ni.3147
The impact of host genetic variation on infection with HIV-1
Abstract
The outcome after infection with the human immunodeficiency virus type 1 (HIV-1) is a complex phenotype determined by interactions among the pathogen, the human host and the surrounding environment. An impact of host genetic variation on HIV-1 susceptibility was identified early in the pandemic, with a major role attributed to the genes encoding class I human leukocyte antigens (HLA) and the chemokine receptor CCR5. Studies using genome-wide data sets have underscored the strength of these associations relative to variants located throughout the rest of the genome. However, the extent to which additional polymorphisms influence HIV-1 disease progression, and how much of the variability in outcome can be attributed to host genetics, remain largely unclear. Here we discuss findings concerning the functional impact of associated variants, outline methods for quantifying the host genetic component and examine how available genome-wide data sets may be leveraged to discover gene variants that affect the outcome of HIV-1 infection.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Figures
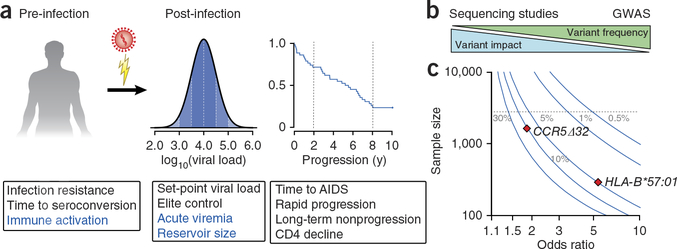
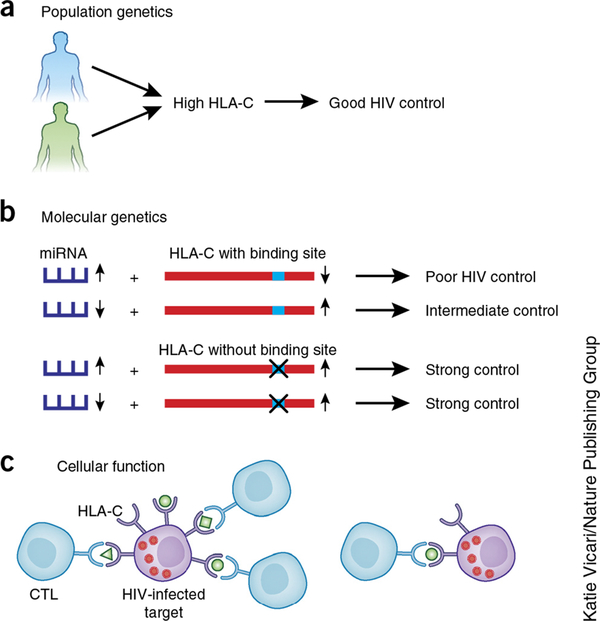
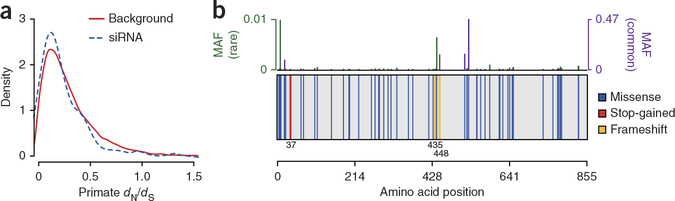
References
-
- Horton RE, McLaren PJ, Fowke K, Kimani J & Ball TB Cohorts for the study of HIV-1-exposed but uninfected individuals: benefits and limitations. J. Infect. Dis 202 (suppl. 3), S377–S381 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
