

Gut microbial succession follows acute secretory diarrhea in humans
- PMID: 25991682
- PMCID: PMC4442136
- DOI: 10.1128/mBio.00381-15
Gut microbial succession follows acute secretory diarrhea in humans
Abstract
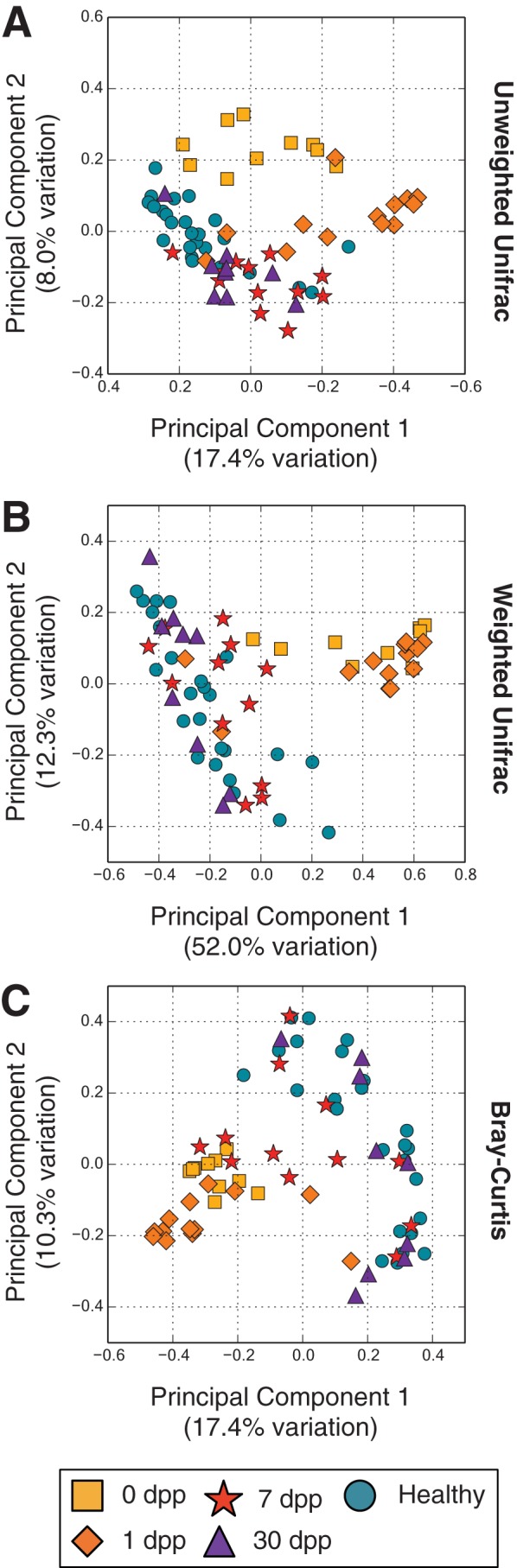
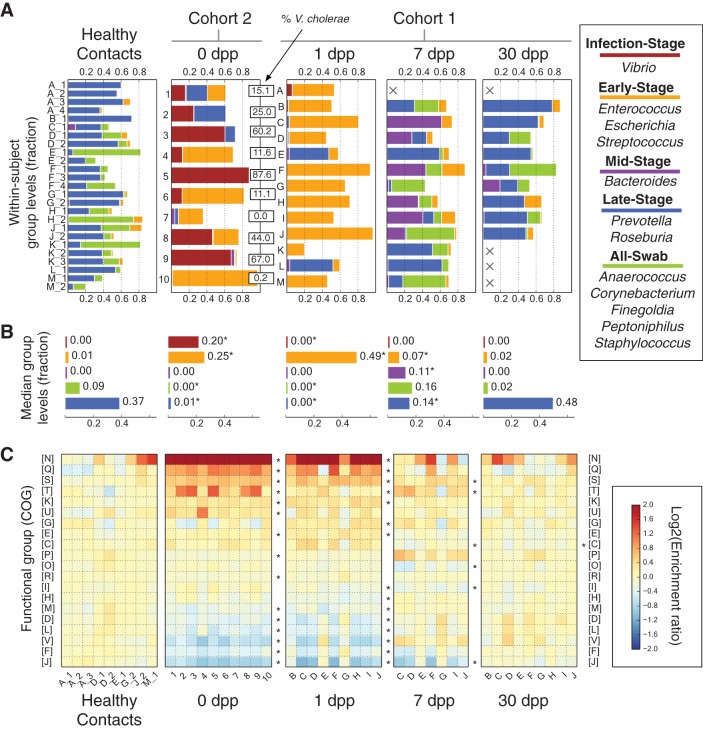
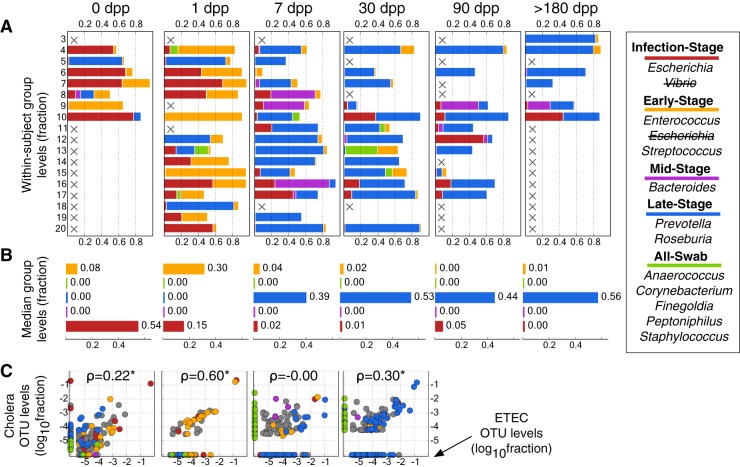
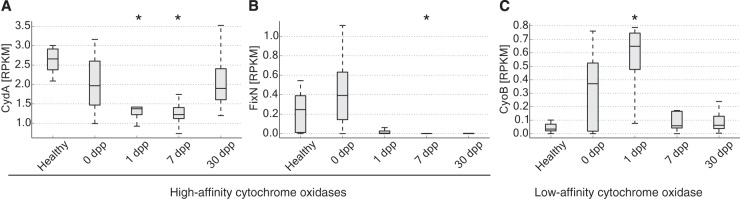
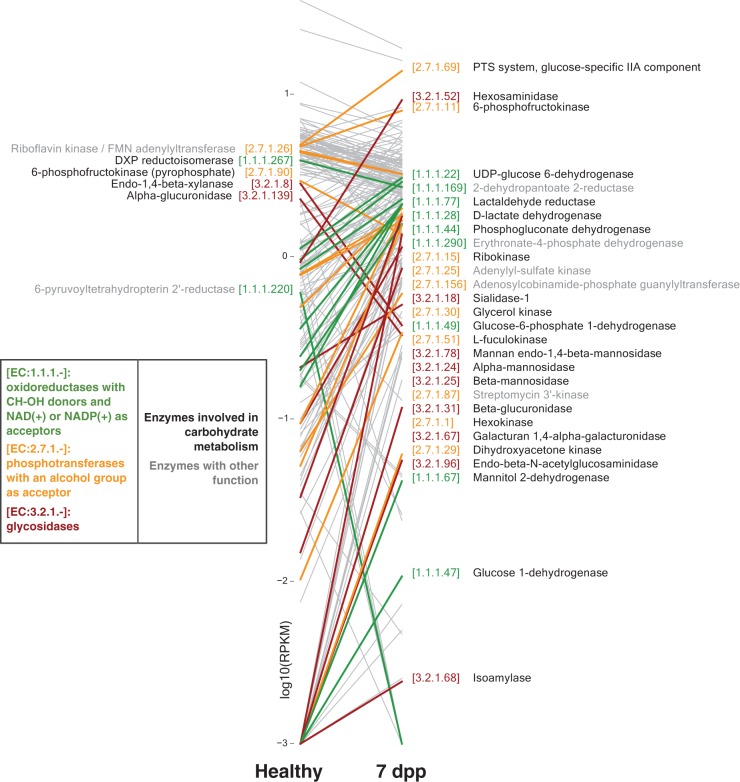
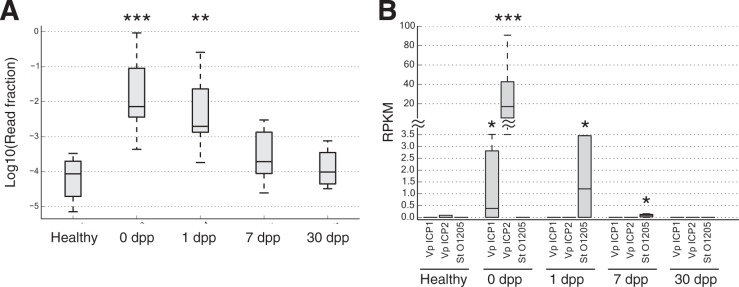
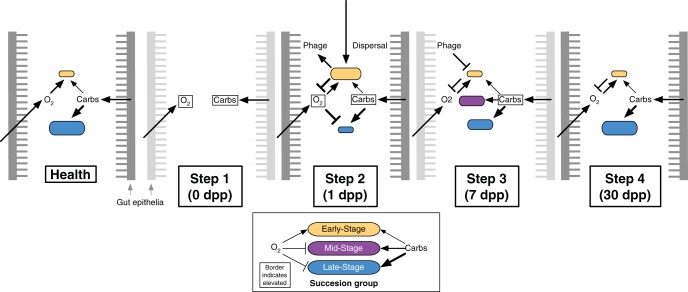
Disability after childhood diarrhea is an important burden on global productivity. Recent studies suggest that gut bacterial communities influence how humans recover from infectious diarrhea, but we still lack extensive data and mechanistic hypotheses for how these bacterial communities respond to diarrheal disease and its treatment. Here, we report that after Vibrio cholerae infection, the human gut microbiota undergoes an orderly and reproducible succession that features transient reversals in relative levels of enteric Bacteroides and Prevotella. Elements of this succession may be a common feature in microbiota recovery from acute secretory diarrhea, as we observed similar successional dynamics after enterotoxigenic Escherichia coli (ETEC) infection. Our metagenomic analyses suggest that multiple mechanisms drive microbial succession after cholera, including bacterial dispersal properties, changing enteric oxygen and carbohydrate levels, and phage dynamics. Thus, gut microbiota recovery after cholera may be predictable at the level of community structure but is driven by a complex set of temporally varying ecological processes. Our findings suggest opportunities for diagnostics and therapies targeting the gut microbiota in humans recovering from infectious diarrhea.
Importance: Disability after diarrhea is a major burden on public health in the developing world. Gut bacteria may affect this recovery, but it remains incompletely understood how resident microbes in the digestive tract respond to diarrheal illness. Here, we observed an orderly and reproducible succession of gut bacterial groups after cholera in humans. Genomic analyses associated the succession with bacterial dispersal in food, an altered microbial environment, and changing phage levels. Our findings suggest that it may one day be feasible to manage resident bacterial populations in the gut after infectious diarrhea.
Copyright © 2015 David et al.
Figures
References
-
- Niehaus MD, Moore SR, Patrick PD, Derr LL, Lorntz B, Lima AA, Guerrant RL. 2002. Early childhood diarrhea is associated with diminished cognitive function 4 to 7-years later in children in a northeast Brazilian shantytown. Am J Trop Med Hyg 66:590–593. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
