

Widespread recombination, reassortment, and transmission of unbalanced compound viral genotypes in natural arenavirus infections
- PMID: 25993603
- PMCID: PMC4438980
- DOI: 10.1371/journal.ppat.1004900
Widespread recombination, reassortment, and transmission of unbalanced compound viral genotypes in natural arenavirus infections
Abstract
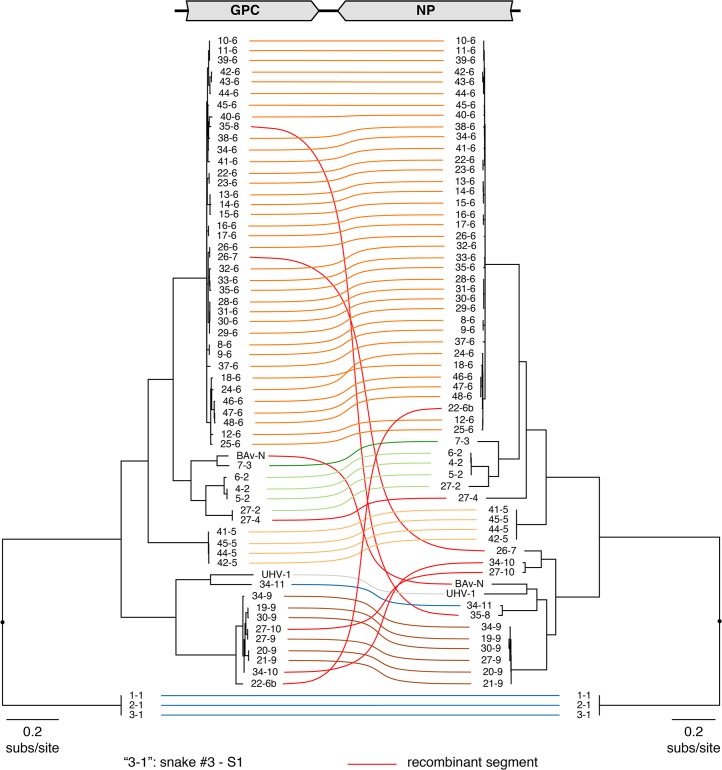
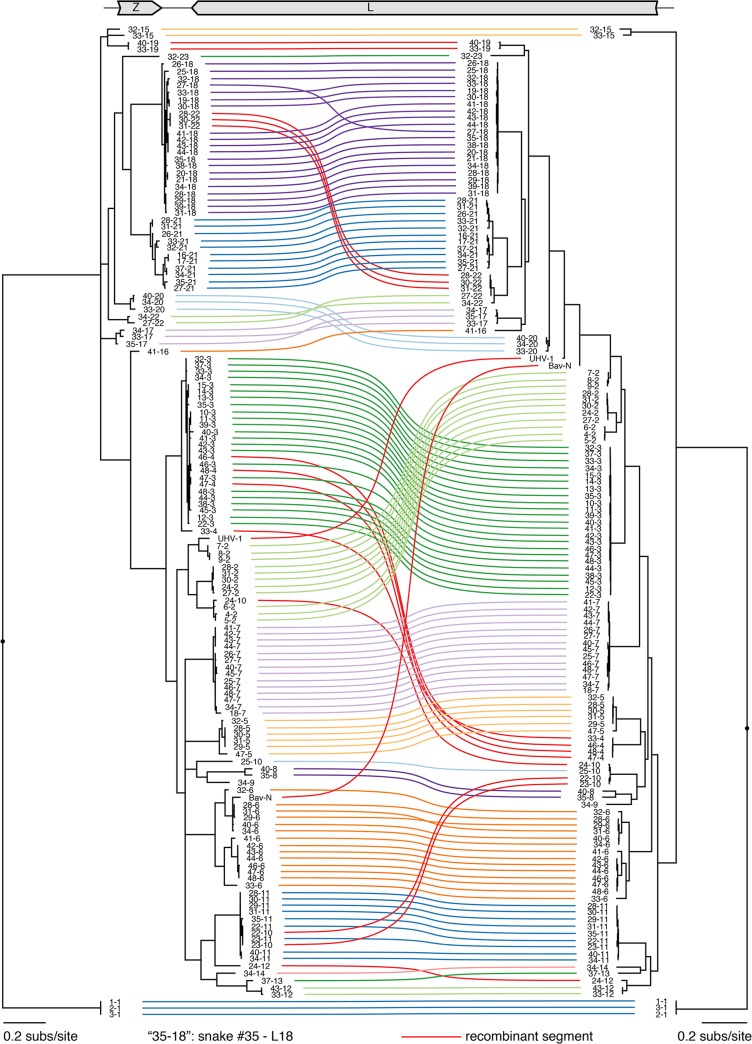
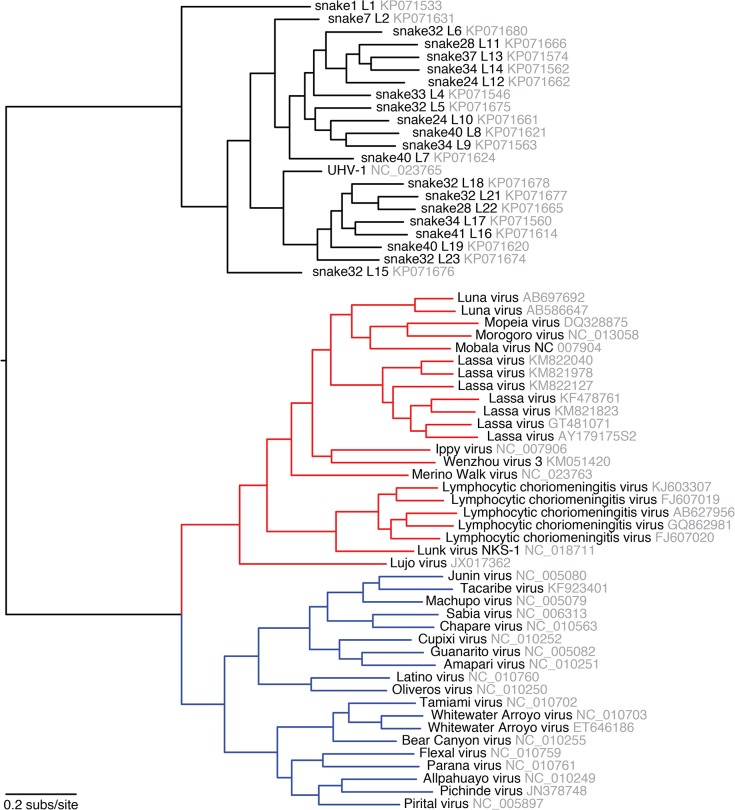
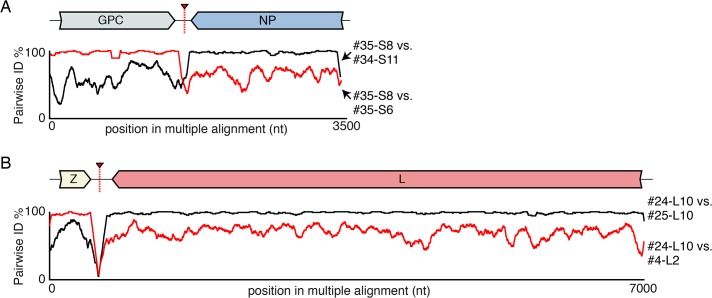
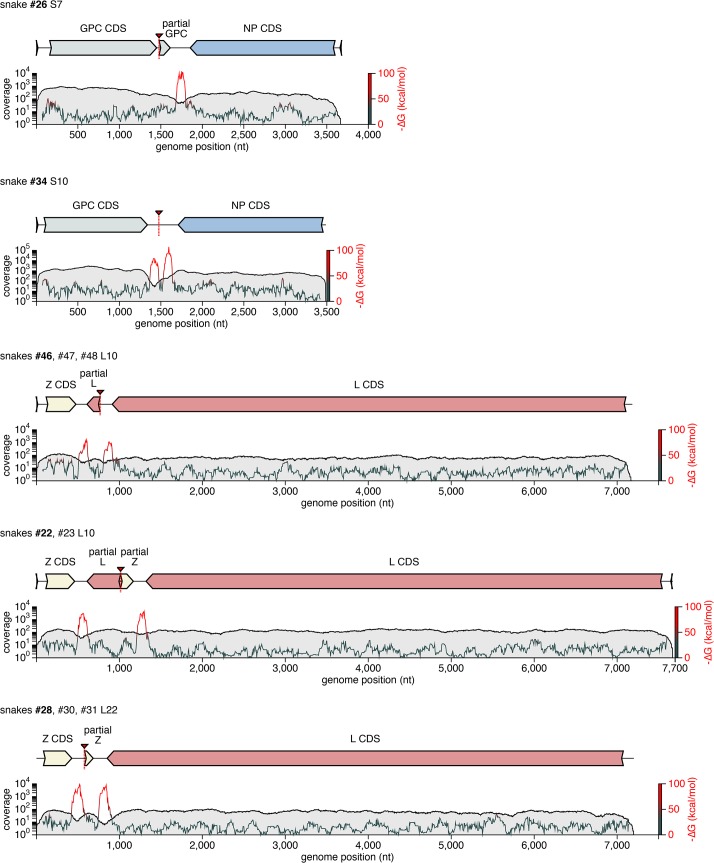
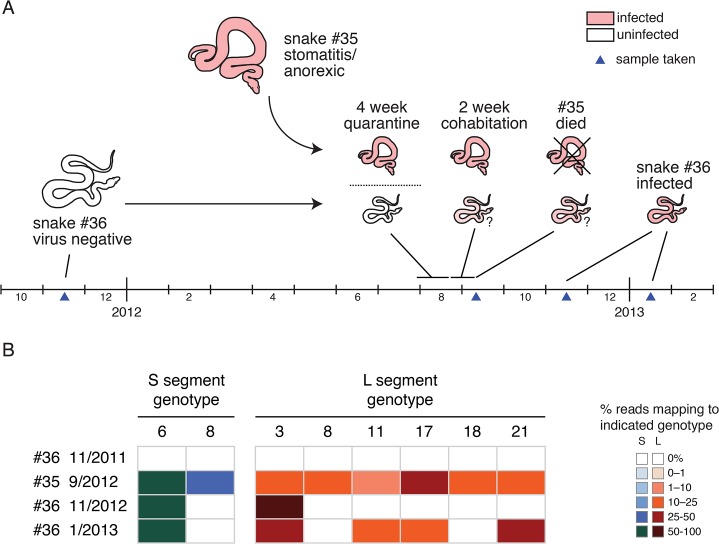
Arenaviruses are one of the largest families of human hemorrhagic fever viruses and are known to infect both mammals and snakes. Arenaviruses package a large (L) and small (S) genome segment in their virions. For segmented RNA viruses like these, novel genotypes can be generated through mutation, recombination, and reassortment. Although it is believed that an ancient recombination event led to the emergence of a new lineage of mammalian arenaviruses, neither recombination nor reassortment has been definitively documented in natural arenavirus infections. Here, we used metagenomic sequencing to survey the viral diversity present in captive arenavirus-infected snakes. From 48 infected animals, we determined the complete or near complete sequence of 210 genome segments that grouped into 23 L and 11 S genotypes. The majority of snakes were multiply infected, with up to 4 distinct S and 11 distinct L segment genotypes in individual animals. This S/L imbalance was typical: in all cases intrahost L segment genotypes outnumbered S genotypes, and a particular S segment genotype dominated in individual animals and at a population level. We corroborated sequencing results by qRT-PCR and virus isolation, and isolates replicated as ensembles in culture. Numerous instances of recombination and reassortment were detected, including recombinant segments with unusual organizations featuring 2 intergenic regions and superfluous content, which were capable of stable replication and transmission despite their atypical structures. Overall, this represents intrahost diversity of an extent and form that goes well beyond what has been observed for arenaviruses or for viruses in general. This diversity can be plausibly attributed to the captive intermingling of sub-clinically infected wild-caught snakes. Thus, beyond providing a unique opportunity to study arenavirus evolution and adaptation, these findings allow the investigation of unintended anthropogenic impacts on viral ecology, diversity, and disease potential.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Holmes EC. Virus Evolution In: D M Knipe, Howley P M, editors. Fields Virology. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2013. pp. 286–313.
-
- Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. Rapid evolution of RNA genomes. Science. 1982;215: 1577–1585. - PubMed
-
- Domingo E, Escarmís C, Sevilla N, Moya A, Elena SF, Quer J, et al. Basic concepts in RNA virus evolution. FASEB J. 1996;10: 859–864. - PubMed
-
- Eigen M. Self organization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971;58: 465–523. - PubMed
Publication types
MeSH terms
Substances
Associated data
- BioProject/PRJNA277217
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
