

Position-specific binding of FUS to nascent RNA regulates mRNA length
- PMID: 25995189
- PMCID: PMC4441052
- DOI: 10.1101/gad.255737.114
Position-specific binding of FUS to nascent RNA regulates mRNA length
Abstract
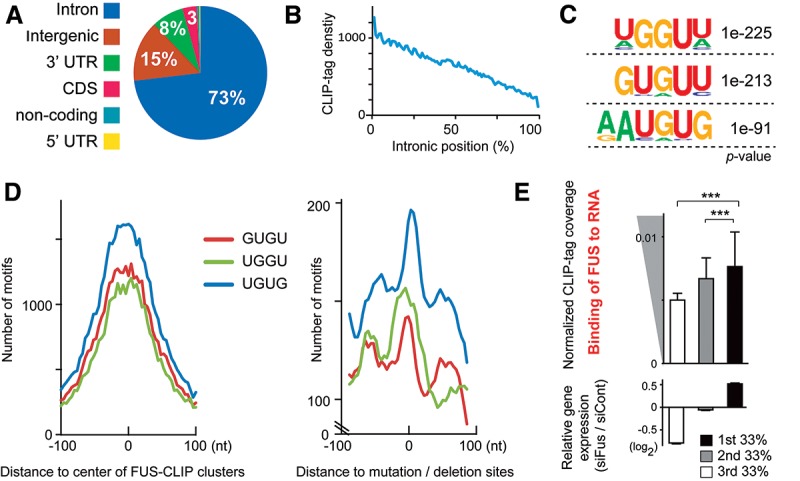
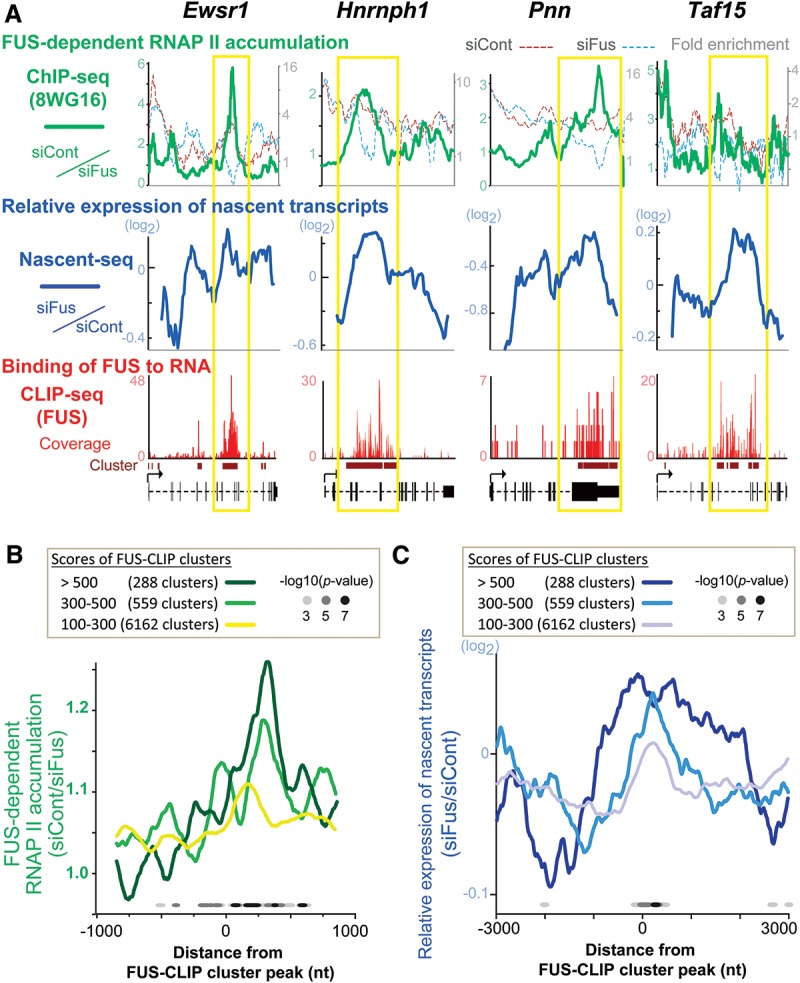
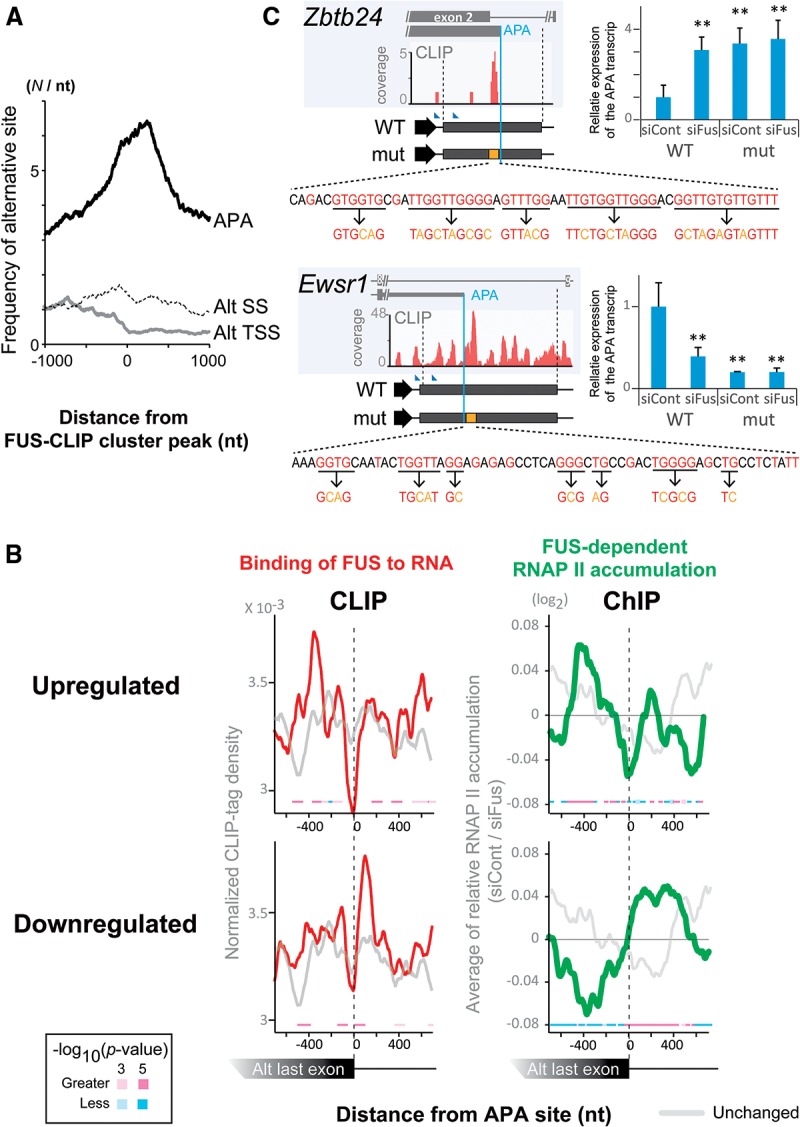
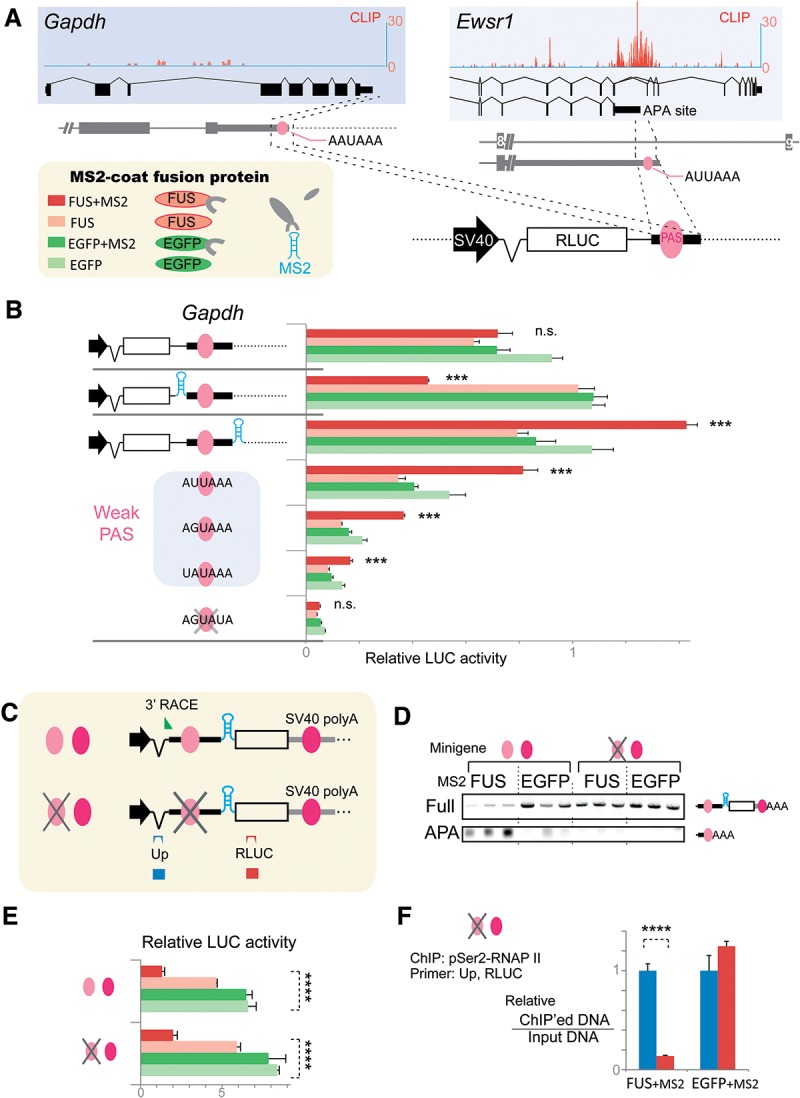
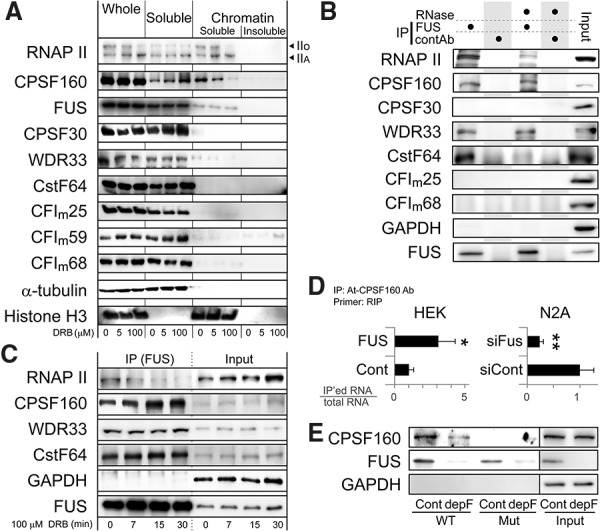
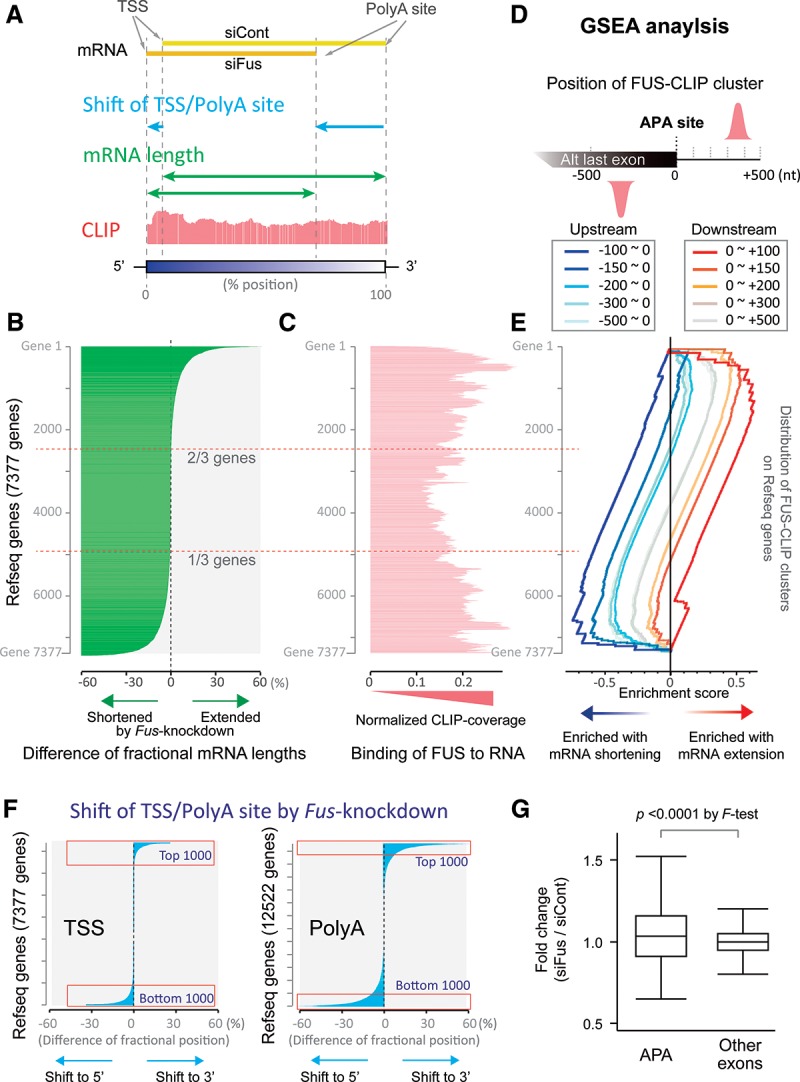
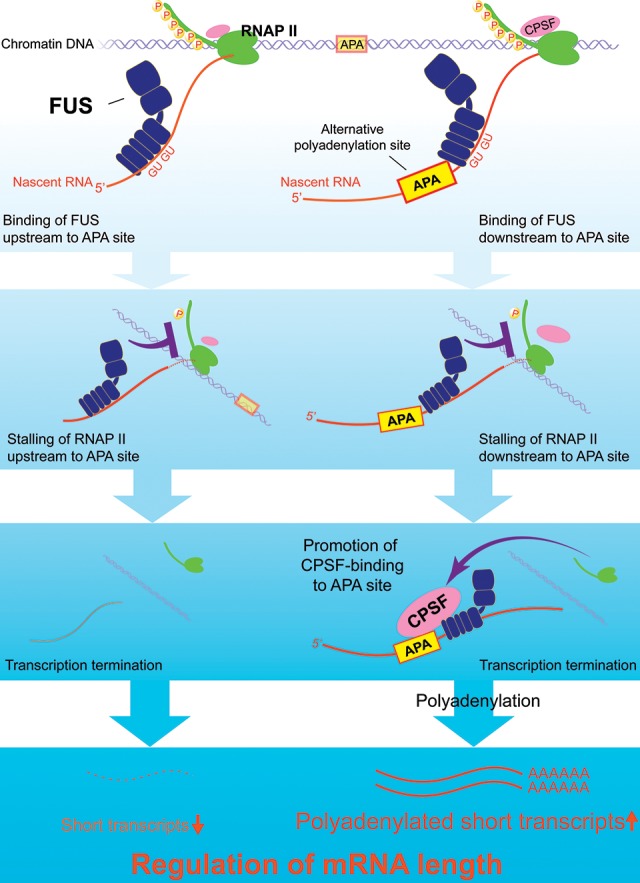
More than half of all human genes produce prematurely terminated polyadenylated short mRNAs. However, the underlying mechanisms remain largely elusive. CLIP-seq (cross-linking immunoprecipitation [CLIP] combined with deep sequencing) of FUS (fused in sarcoma) in neuronal cells showed that FUS is frequently clustered around an alternative polyadenylation (APA) site of nascent RNA. ChIP-seq (chromatin immunoprecipitation [ChIP] combined with deep sequencing) of RNA polymerase II (RNAP II) demonstrated that FUS stalls RNAP II and prematurely terminates transcription. When an APA site is located upstream of an FUS cluster, FUS enhances polyadenylation by recruiting CPSF160 and up-regulates the alternative short transcript. In contrast, when an APA site is located downstream from an FUS cluster, polyadenylation is not activated, and the RNAP II-suppressing effect of FUS leads to down-regulation of the alternative short transcript. CAGE-seq (cap analysis of gene expression [CAGE] combined with deep sequencing) and PolyA-seq (a strand-specific and quantitative method for high-throughput sequencing of 3' ends of polyadenylated transcripts) revealed that position-specific regulation of mRNA lengths by FUS is operational in two-thirds of transcripts in neuronal cells, with enrichment in genes involved in synaptic activities.
Keywords: CLIP; FUS; RNA polymerase II; alternative polyadenylation; mRNA length.
© 2015 Masuda et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell 45: 158–170. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous