

Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma
- PMID: 25995224
- PMCID: PMC4634889
- DOI: 10.1126/scitranslmed.aaa7014
Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma
Abstract
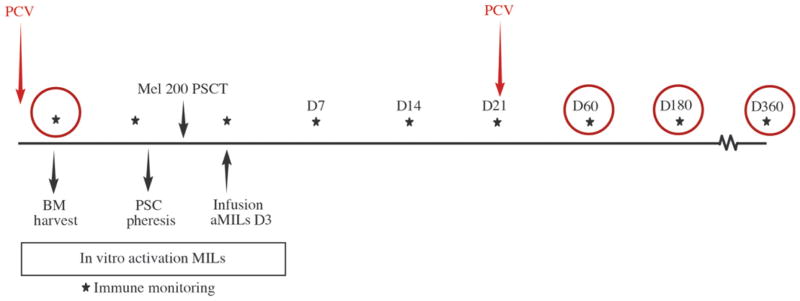
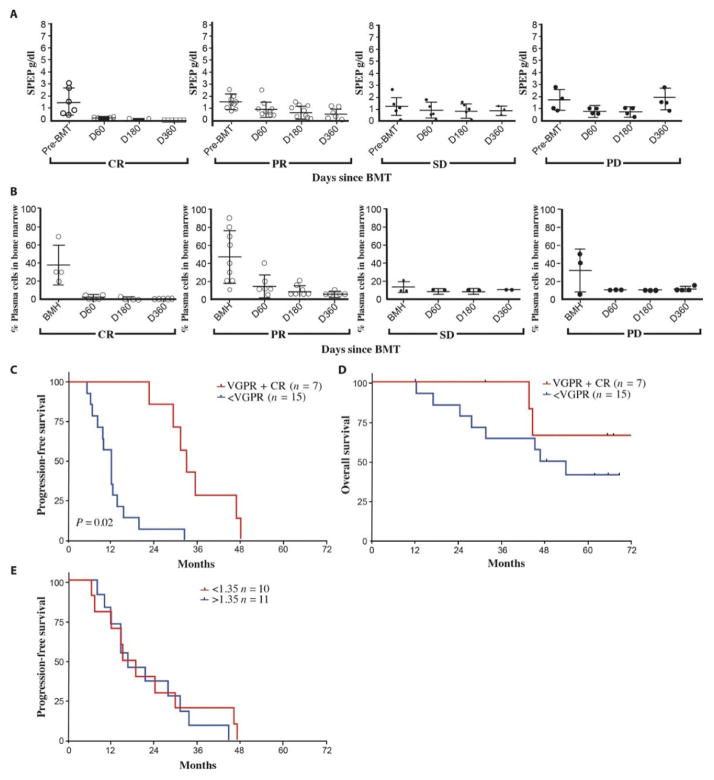
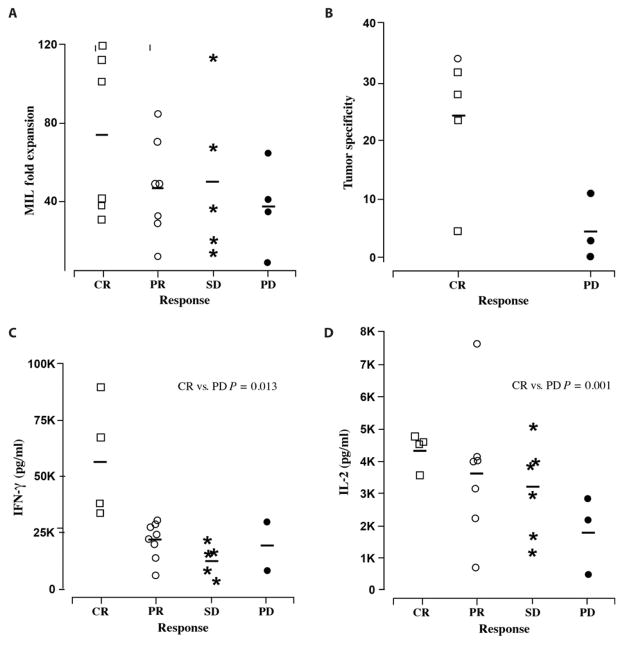
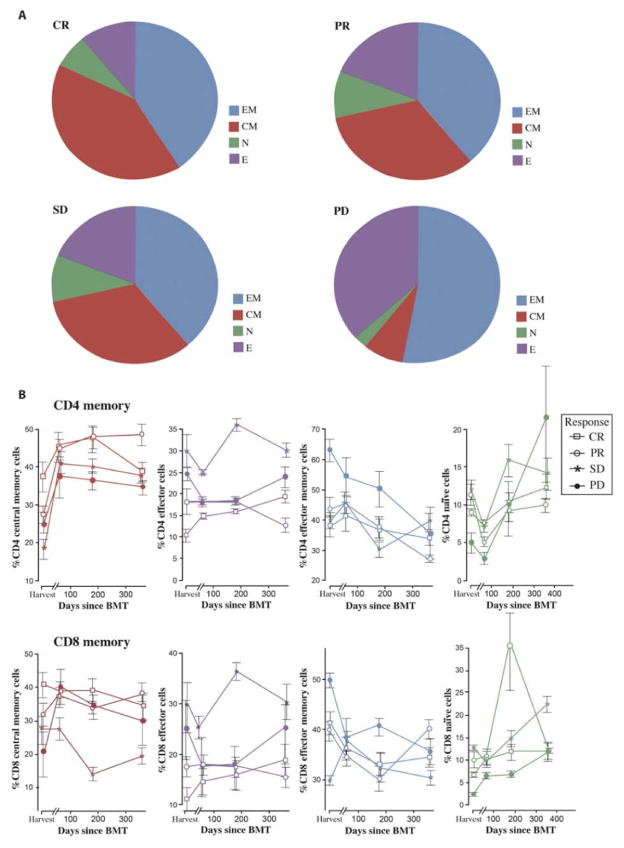
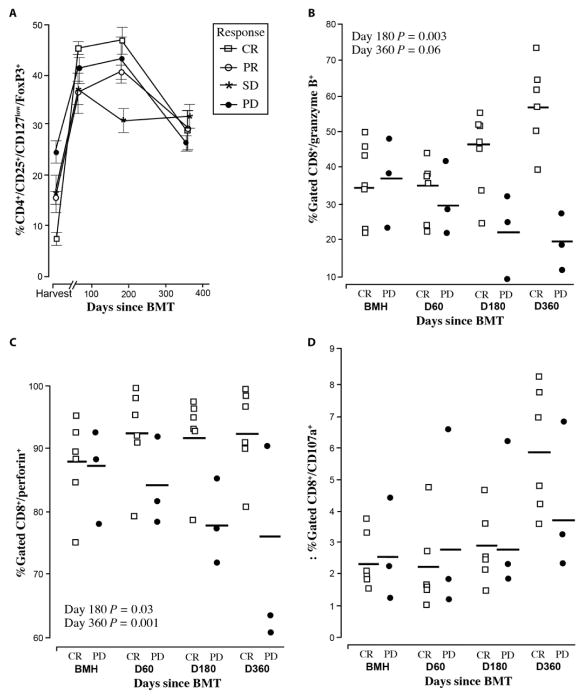
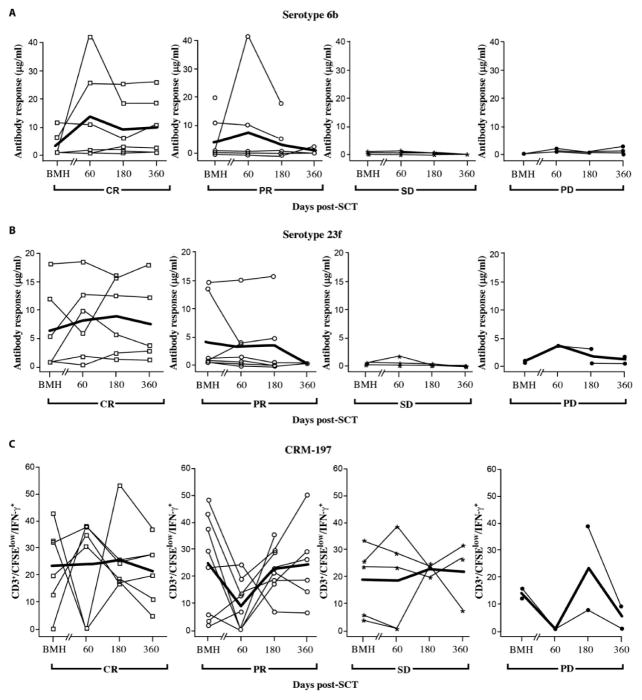
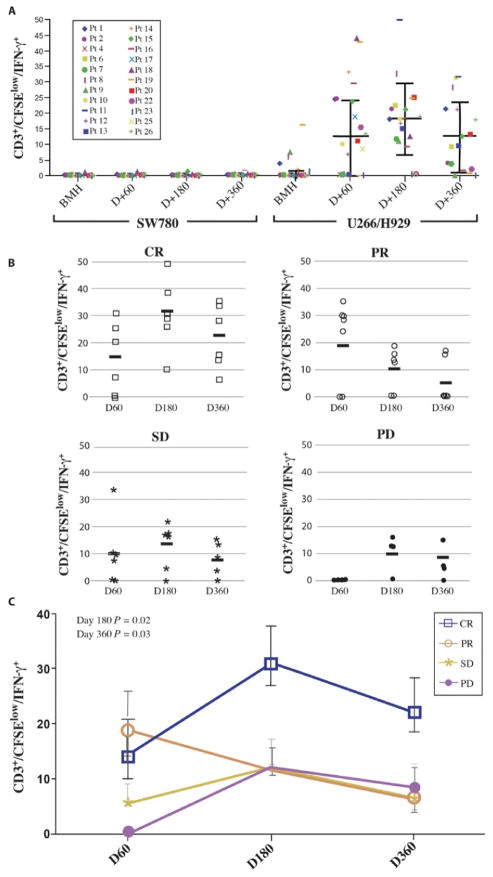
Successful adoptive T cell therapy (ACT) requires the ability to activate tumor-specific T cells with the ability to traffic to the tumor site and effectively kill their target as well as persist over time. We hypothesized that ACT using marrow-infiltrating lymphocytes (MILs) in multiple myeloma (MM) could impart greater antitumor immunity in that they were obtained from the tumor microenvironment. We describe the results from the first clinical trial using MILs in MM. Twenty-five patients with either newly diagnosed or relapsed disease had their MILs harvested, activated and expanded, and subsequently infused on the third day after myeloablative therapy. Cells were obtained and adequately expanded in all patients with anti-CD3/CD28 beads plus interleukin-2, and a median of 9.5 × 10(8) MILs were infused. Factors indicative of response to MIL ACT included (i) the presence of measurable myeloma-specific activity of the ex vivo expanded product, (ii) low endogenous bone marrow T cell interferon-γ production at baseline, (iii) a CD8(+) central memory phenotype at baseline, and (iv) the generation and persistence of myeloma-specific immunity in the bone marrow at 1 year after ACT. Achieving at least a 90% reduction in disease burden significantly increased the progression-free survival (25.1 months versus 11.8 months; P = 0.01). This study demonstrates the feasibility and efficacy of MILs as a form of ACT with applicability across many hematologic malignancies and possibly solid tumors infiltrating the bone marrow.
Trial registration: ClinicalTrials.gov NCT00566098.
Copyright © 2015, American Association for the Advancement of Science.
Conflict of interest statement
Figures
References
-
- Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, Casassus P, Maisonneuve H, Facon T, Ifrah N, Payen C, Bataille R. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335:91–97. - PubMed
-
- Borrello I, Sotomayor EM, Rattis FM, Cooke SK, Gu L, Levitsky HI. Sustaining the graft-versus-tumor effect through posttransplant immunization with granulocyte-macrophage colony-stimulating factor (GM-CSF)–producing tumor vaccines. Blood. 2000;95:3011–3019. - PubMed
-
- Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, Veloso E, Zheng Z, Westphal S, Mair R, Chi N, Ratterree B, Pochran MF, Natt S, Hinkle J, Sickles C, Sohal A, Ruehle K, Lynch C, Zhang L, Porter DL, Luger S, Guo C, Fang HB, Blackwelder W, Hankey K, Mann D, Edelman R, Frasch C, Levine BL, Cross A, June CH. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–1237. - PubMed
-
- Porrata LF, Gertz MA, Inwards DJ, Litzow MR, Lacy MQ, Tefferi A, Gastineau DA, Dispenzieri A, Ansell SM, Micallef IN, Geyer SM, Markovic SN. Early lymphocyte recovery predicts superior survival after autologous hematopoietic stem cell transplantation in multiple myeloma or non-Hodgkin lymphoma. Blood. 2001;98:579–585. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
