

FUSE Binding Protein 1 Facilitates Persistent Hepatitis C Virus Replication in Hepatoma Cells by Regulating Tumor Suppressor p53
- PMID: 25995247
- PMCID: PMC4505638
- DOI: 10.1128/JVI.00729-15
FUSE Binding Protein 1 Facilitates Persistent Hepatitis C Virus Replication in Hepatoma Cells by Regulating Tumor Suppressor p53
Erratum in
-
Correction for Dixit et al., "FUSE Binding Protein 1 Facilitates Persistent Hepatitis C Virus Replication in Hepatoma Cells by Regulating Tumor Suppressor p53".J Virol. 2017 Nov 14;91(23):e01609-17. doi: 10.1128/JVI.01609-17. Print 2017 Dec 1. J Virol. 2017. PMID: 29138336 Free PMC article. No abstract available.
Abstract
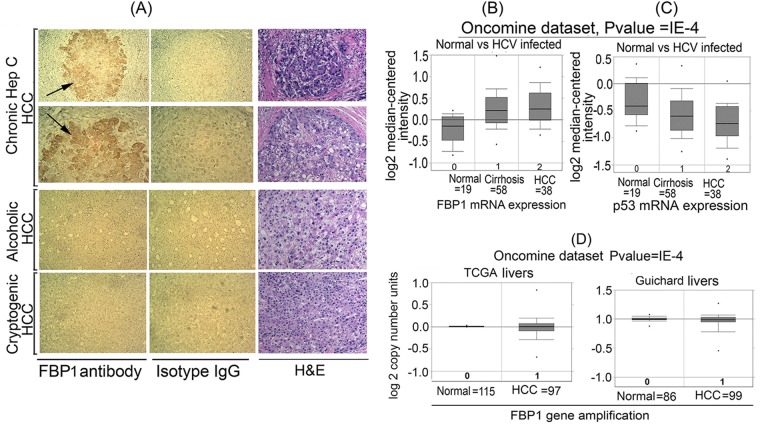
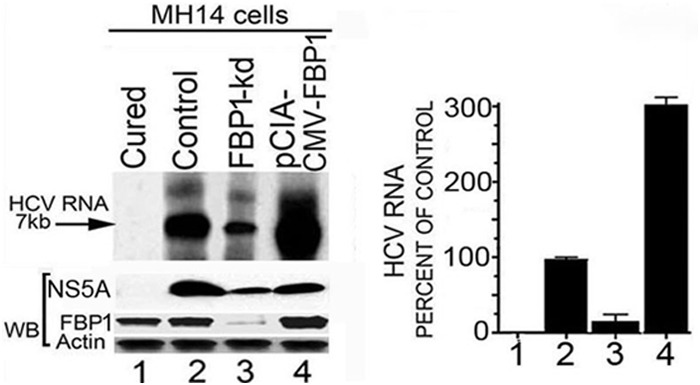
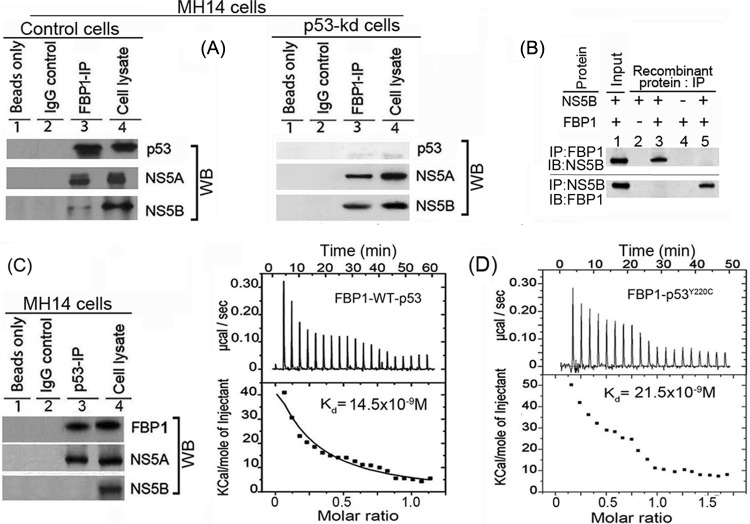
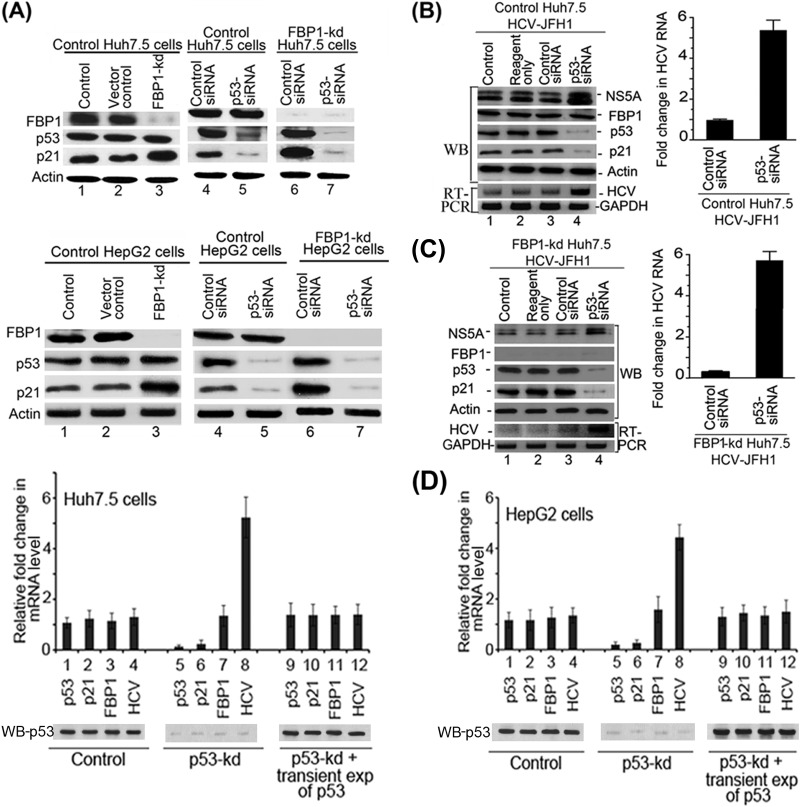
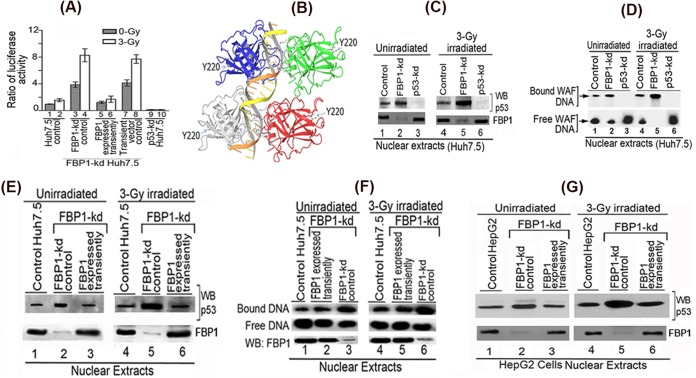
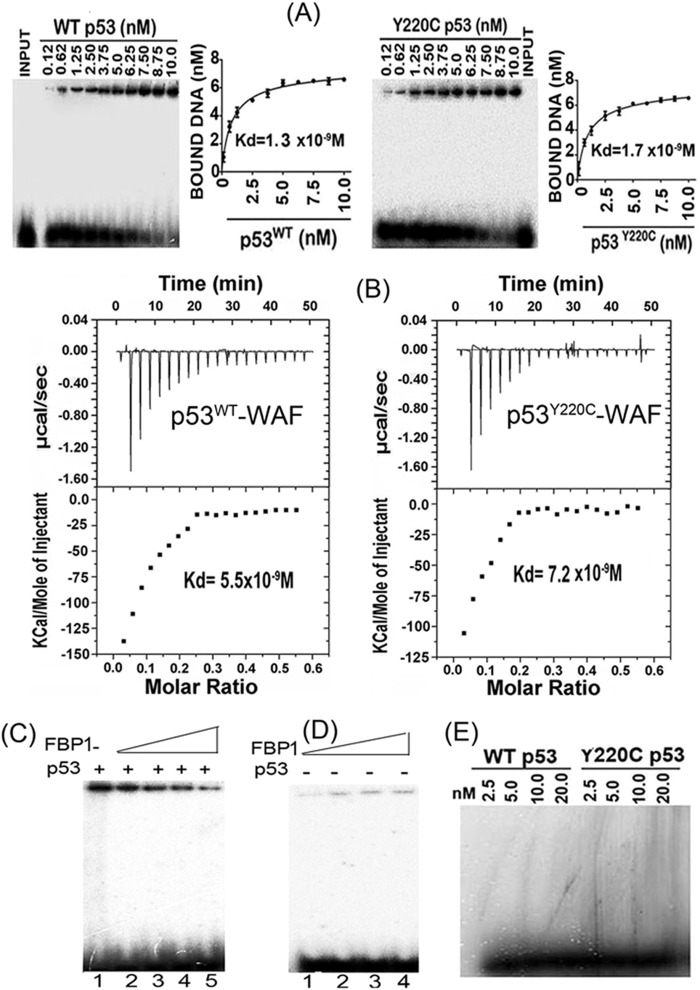
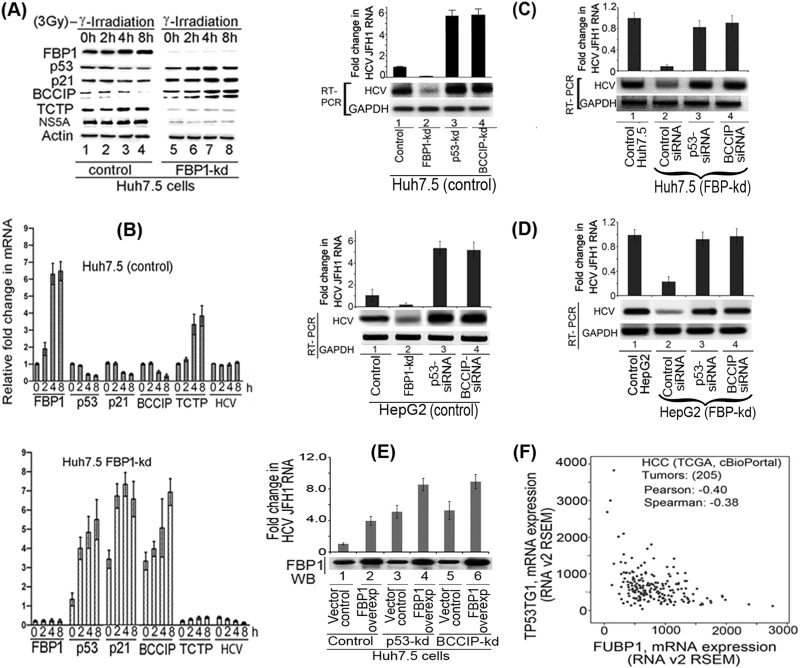
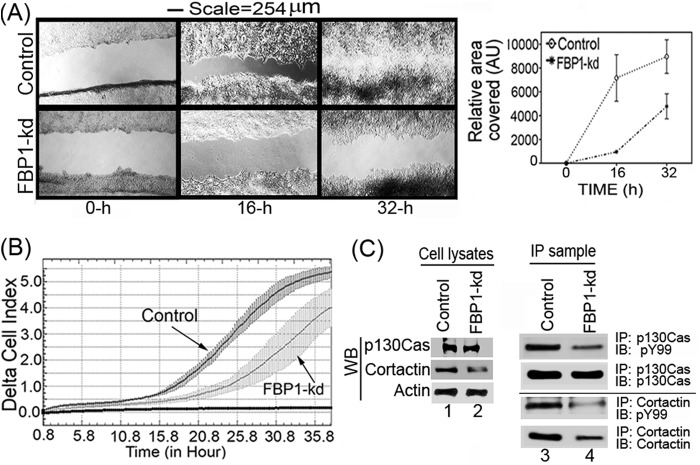
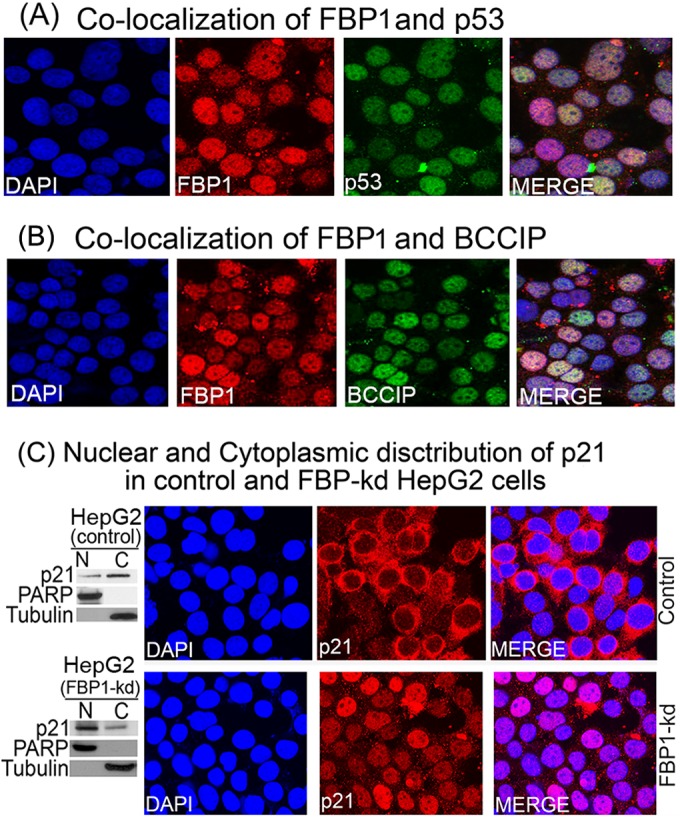
Hepatitis C virus (HCV) is a leading cause of chronic hepatitis C (CHC), liver cirrhosis, and hepatocellular carcinoma (HCC). Immunohistochemistry of archived HCC tumors showed abundant FBP1 expression in HCC tumors with the CHC background. Oncomine data analysis of normal versus HCC tumors with the CHC background indicated a 4-fold increase in FBP1 expression with a concomitant 2.5-fold decrease in the expression of p53. We found that FBP1 promotes HCV replication by inhibiting p53 and regulating BCCIP and TCTP, which are positive and negative regulators of p53, respectively. The severe inhibition of HCV replication in FBP1-knockdown Huh7.5 cells was restored to a normal level by downregulation of either p53 or BCCIP. Although p53 in Huh7.5 cells is transcriptionally inactive as a result of Y220C mutation, we found that the activation and DNA binding ability of Y220C p53 were strongly suppressed by FBP1 but significantly activated upon knockdown of FBP1. Transient expression of FBP1 in FBP1 knockdown cells fully restored the control phenotype in which the DNA binding ability of p53 was strongly suppressed. Using electrophoretic mobility shift assay (EMSA) and isothermal titration calorimetry (ITC), we found no significant difference in in vitro target DNA binding affinity of recombinant wild-type p53 and its Y220C mutant p53. However, in the presence of recombinant FBP1, the DNA binding ability of p53 is strongly inhibited. We confirmed that FBP1 downregulates BCCIP, p21, and p53 and upregulates TCTP under radiation-induced stress. Since FBP1 is overexpressed in most HCC tumors with an HCV background, it may have a role in promoting persistent virus infection and tumorigenesis.
Importance: It is our novel finding that FUSE binding protein 1 (FBP1) strongly inhibits the function of tumor suppressor p53 and is an essential host cell factor required for HCV replication. Oncomine data analysis of a large number of samples has revealed that overexpression of FBP1 in most HCC tumors with chronic hepatitis C is significantly linked with the decreased expression level of p53. The most significant finding is that FBP1 not only physically interacts with p53 and interferes with its binding to the target DNA but also functions as a negative regulator of p53 under cellular stress. FBP1 is barely detectable in normal differentiated cells; its overexpression in HCC tumors with the CHC background suggests that FBP1 has an important role in promoting HCV infection and HCC tumors by suppressing p53.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Fuse binding protein antagonizes the transcription activity of tumor suppressor protein p53.BMC Cancer. 2014 Dec 8;14:925. doi: 10.1186/1471-2407-14-925. BMC Cancer. 2014. PMID: 25487856 Free PMC article.
-
Hepatitis C virus core protein inhibits tumor suppressor protein promyelocytic leukemia function in human hepatoma cells.Cancer Res. 2005 Dec 1;65(23):10830-7. doi: 10.1158/0008-5472.CAN-05-0880. Cancer Res. 2005. PMID: 16322229
-
Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma.Cancer Res. 2016 Jun 1;76(11):3265-76. doi: 10.1158/0008-5472.CAN-15-2601. Epub 2016 Apr 6. Cancer Res. 2016. PMID: 27197151
-
Hepatocellular carcinoma: molecular interactions between hepatitis C virus and p53 in hepatocarcinogenesis.Expert Rev Mol Med. 2003 Nov 19;5(28):1-16. doi: 10.1017/S1462399403006926. Expert Rev Mol Med. 2003. PMID: 14987403 Review.
-
[Molecular genetic and epigenetic mechanisms of hepatocarcinogenesis].Ai Zheng. 2005 Jun;24(6):757-68. Ai Zheng. 2005. PMID: 15946497 Review. Chinese.
Cited by
-
Cortactin Interacts with Hepatitis C Virus Core and NS5A Proteins: Implications for Virion Assembly.J Virol. 2020 Sep 15;94(19):e01306-20. doi: 10.1128/JVI.01306-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32727880 Free PMC article.
-
The role of MDM2-p53 axis dysfunction in the hepatocellular carcinoma transformation.Cell Death Discov. 2020 Jun 19;6:53. doi: 10.1038/s41420-020-0287-y. eCollection 2020. Cell Death Discov. 2020. PMID: 32595984 Free PMC article. Review.
-
Adenosine deaminase acting on RNA-1 (ADAR1) inhibits hepatitis B virus (HBV) replication by enhancing microRNA-122 processing.J Biol Chem. 2019 Sep 20;294(38):14043-14054. doi: 10.1074/jbc.RA119.007970. Epub 2019 Jul 30. J Biol Chem. 2019. PMID: 31366735 Free PMC article.
-
FUSE binding protein FUBP3 is a potent regulator in Japanese encephalitis virus infection.Virol J. 2021 Nov 18;18(1):224. doi: 10.1186/s12985-021-01697-8. Virol J. 2021. PMID: 34794468 Free PMC article.
-
Staufen1 promotes HCV replication by inhibiting protein kinase R and transporting viral RNA to the site of translation and replication in the cells.Nucleic Acids Res. 2016 Jun 20;44(11):5271-87. doi: 10.1093/nar/gkw312. Epub 2016 Apr 22. Nucleic Acids Res. 2016. PMID: 27106056 Free PMC article.
References
-
- Choo QL, Weiner AJ, Overby LR, Kuo G, Houghton M, Bradley DW. 1990. Hepatitis C virus: the major causative agent of viral non-A, non-B hepatitis. Br Med Bull 46:423–441. - PubMed
-
- Denniston MM, Klevens RM, McQuillan GM, Jiles RB. 2012. Awareness of infection, knowledge of hepatitis C, and medical follow-up among individuals testing positive for hepatitis C: National Health and Nutrition Examination Survey 2001-2008. Hepatology 55:1652–1661. doi:10.1002/hep.25556. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
