

Multiplexed peptide analysis using data-independent acquisition and Skyline
- PMID: 25996789
- PMCID: PMC5127711
- DOI: 10.1038/nprot.2015.055
Multiplexed peptide analysis using data-independent acquisition and Skyline
Abstract
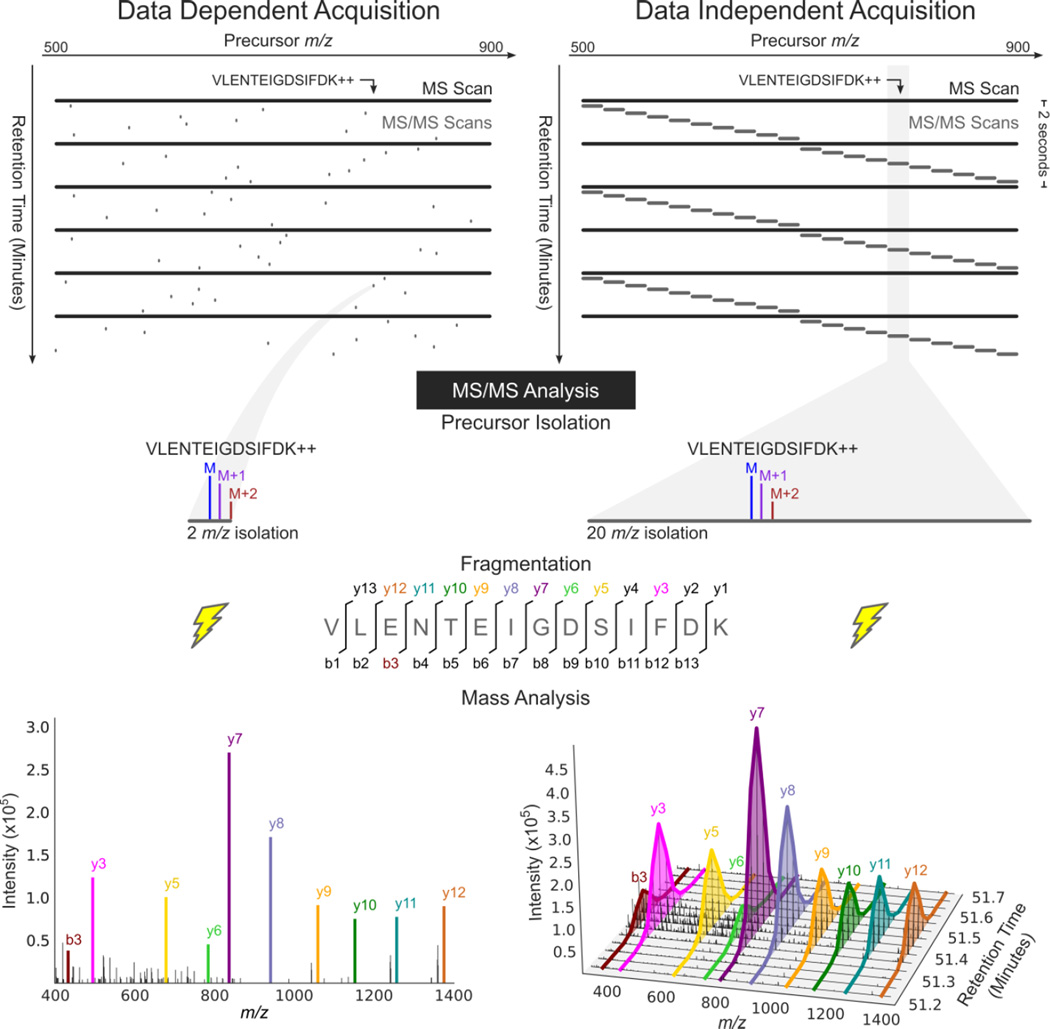
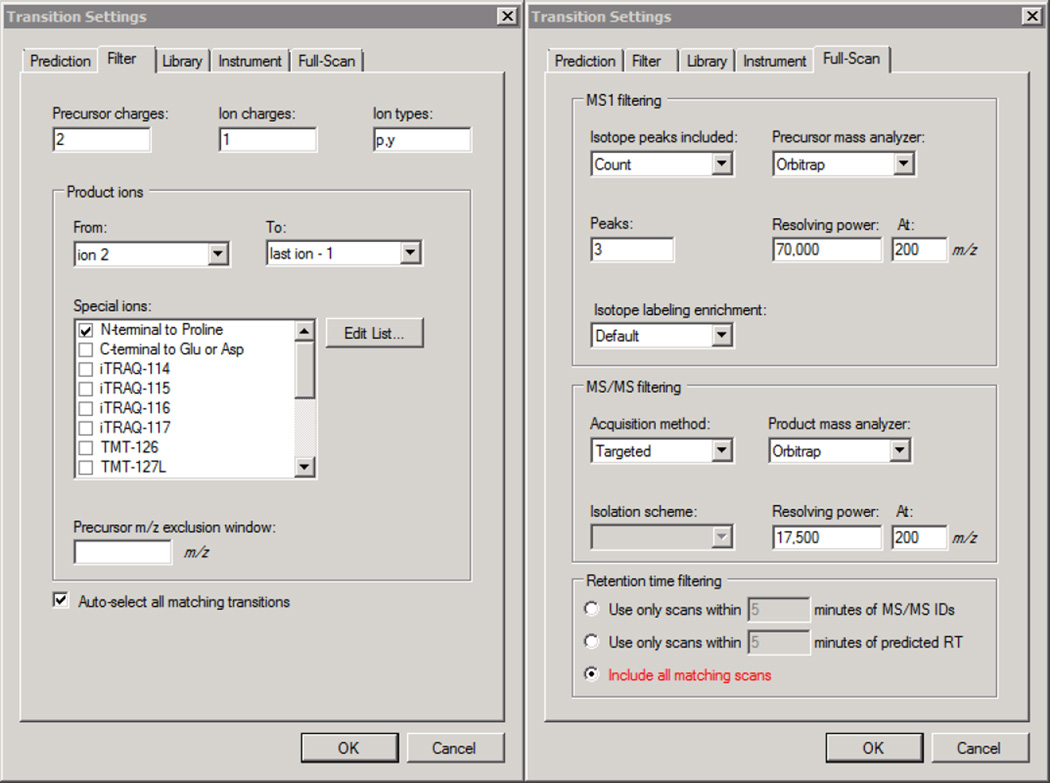
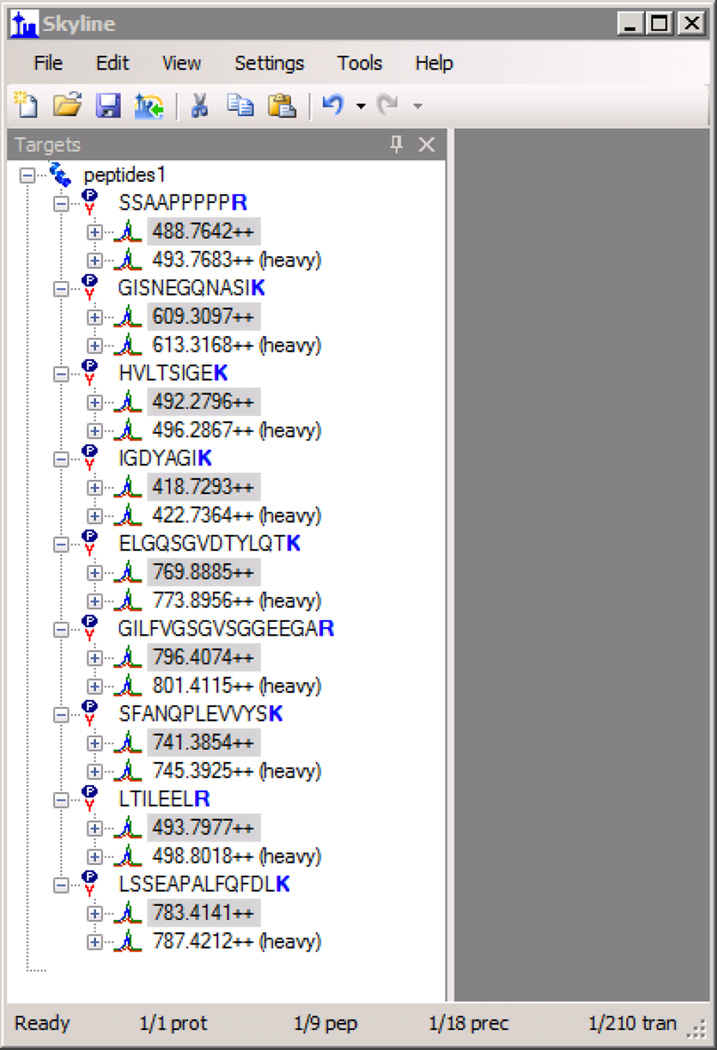
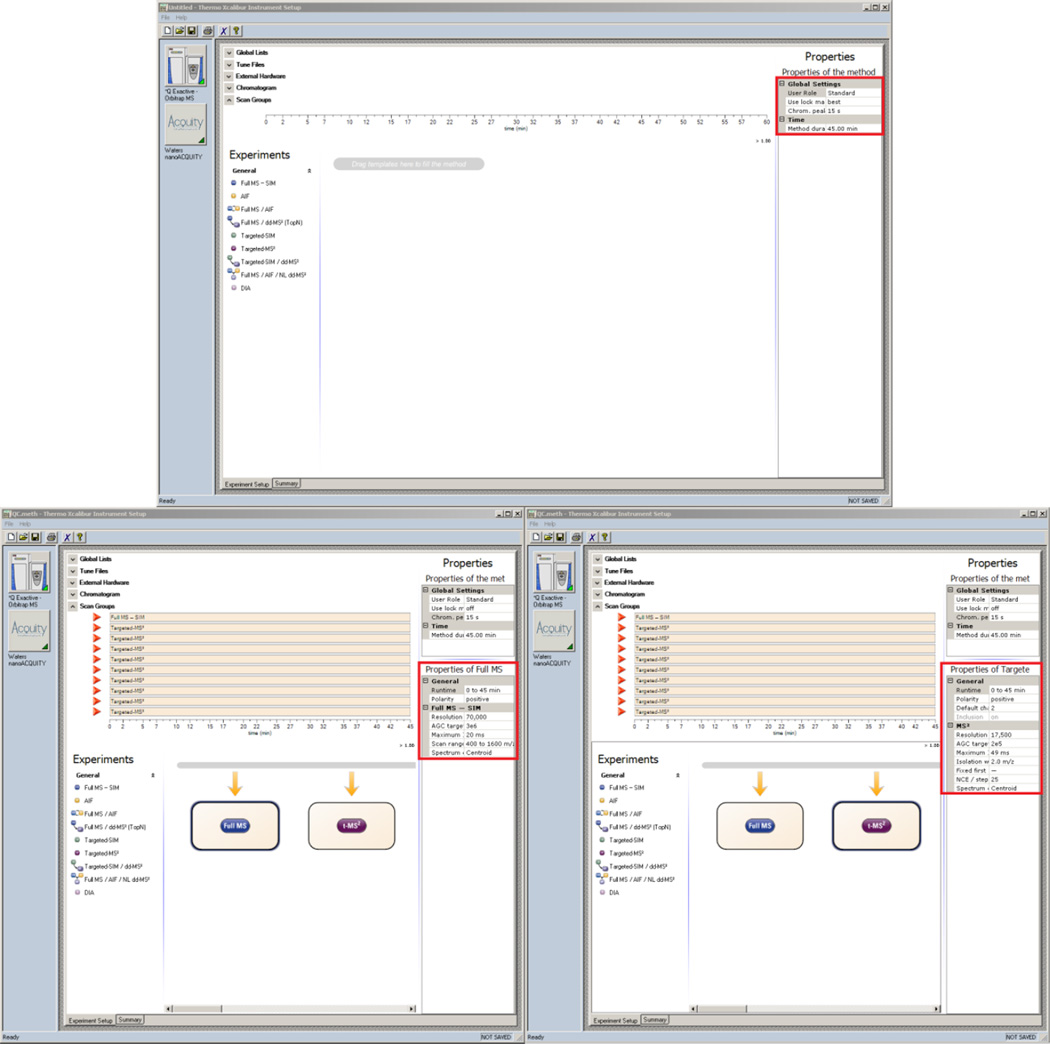
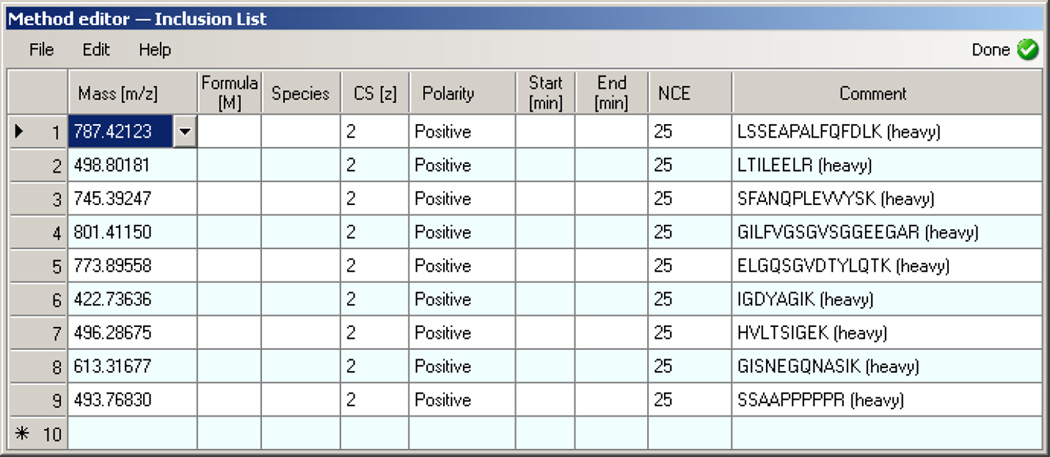
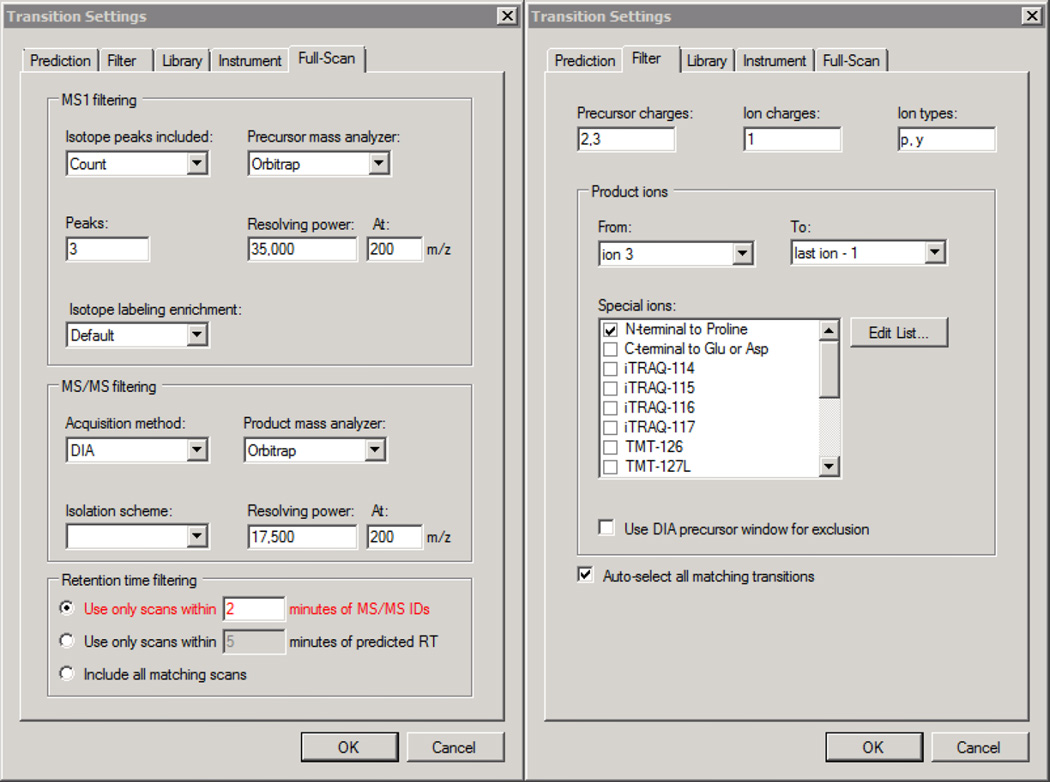
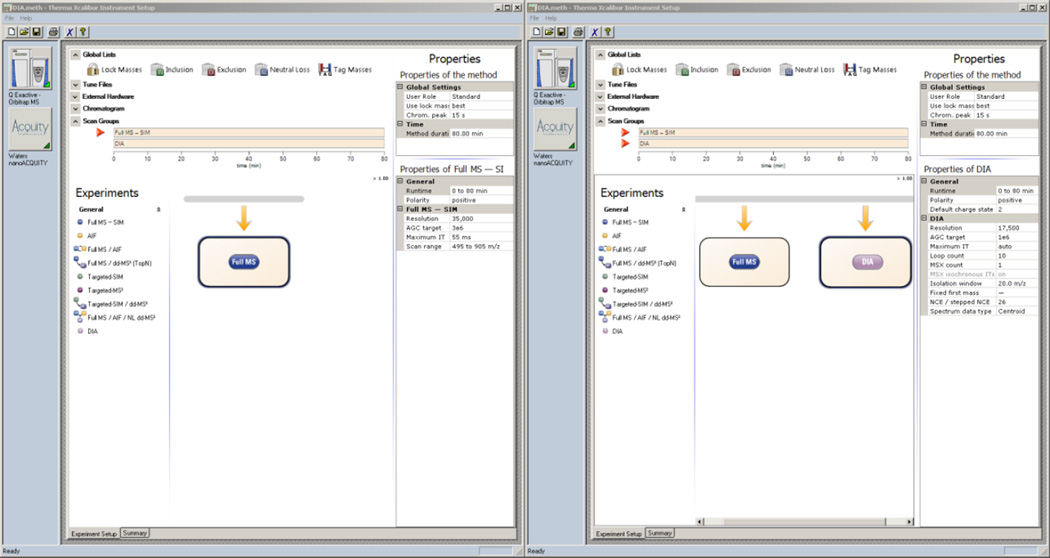
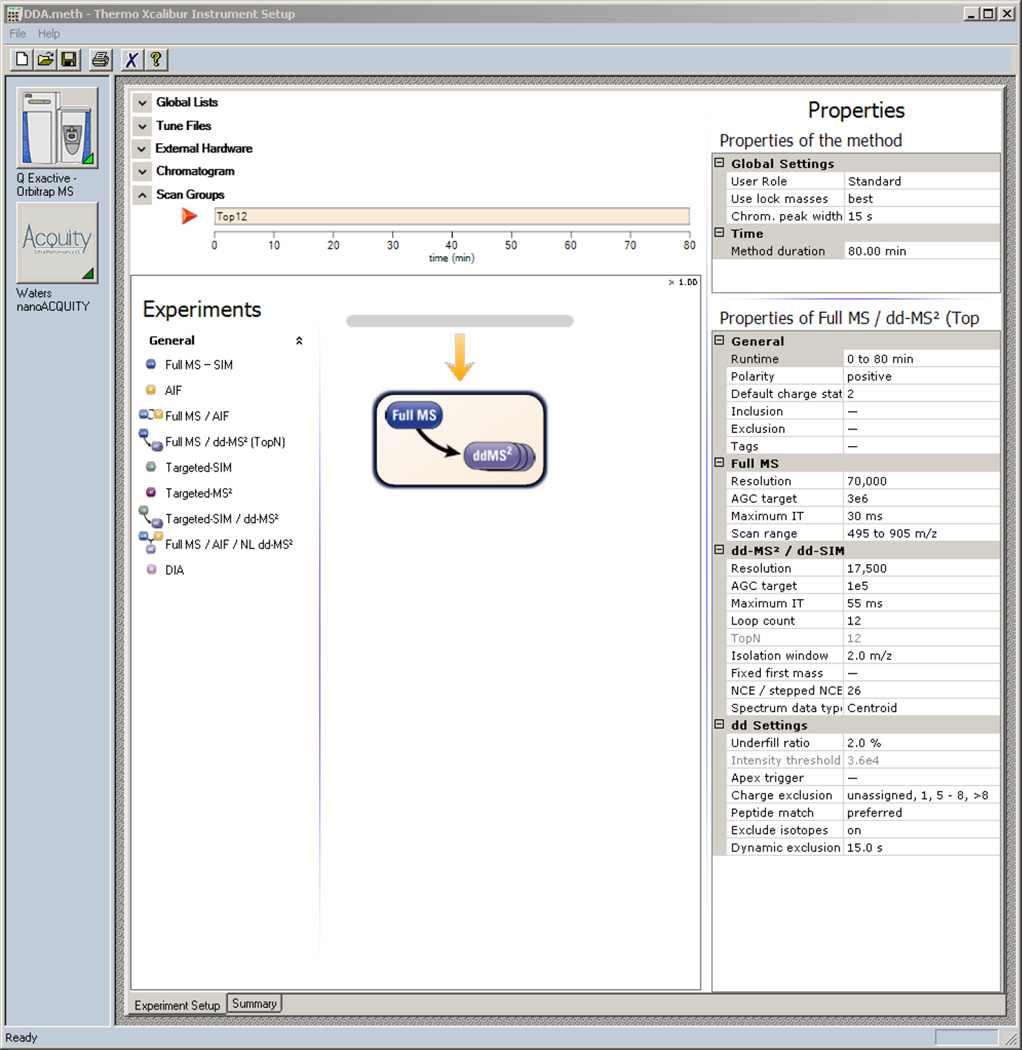
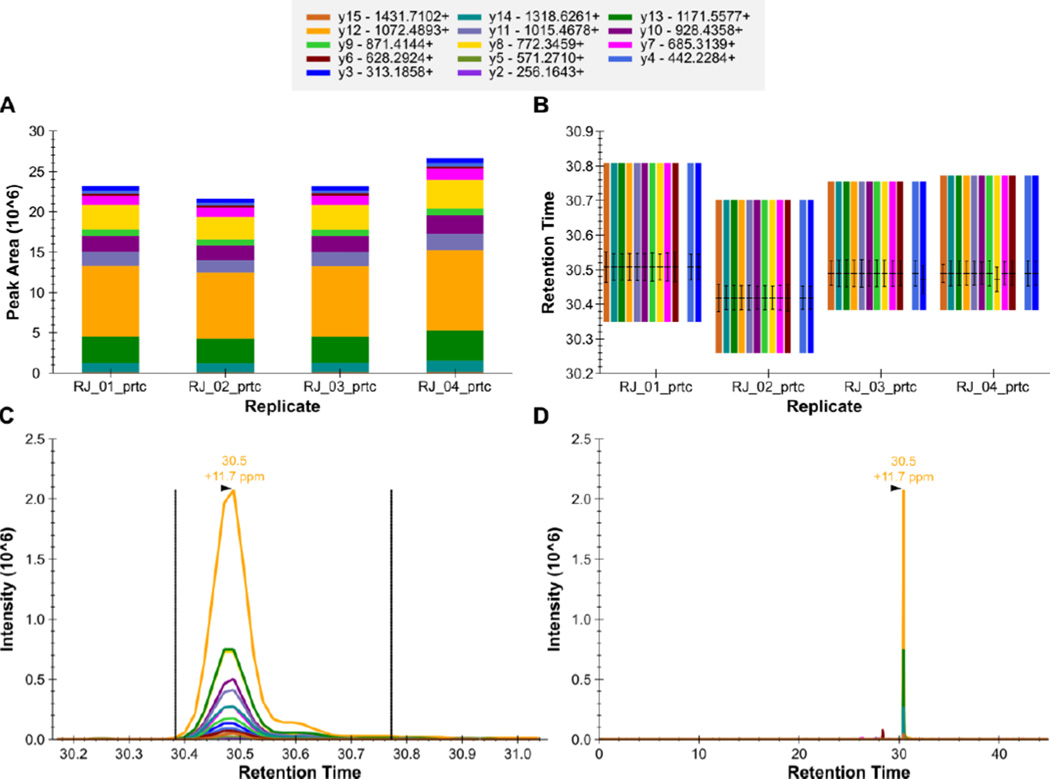
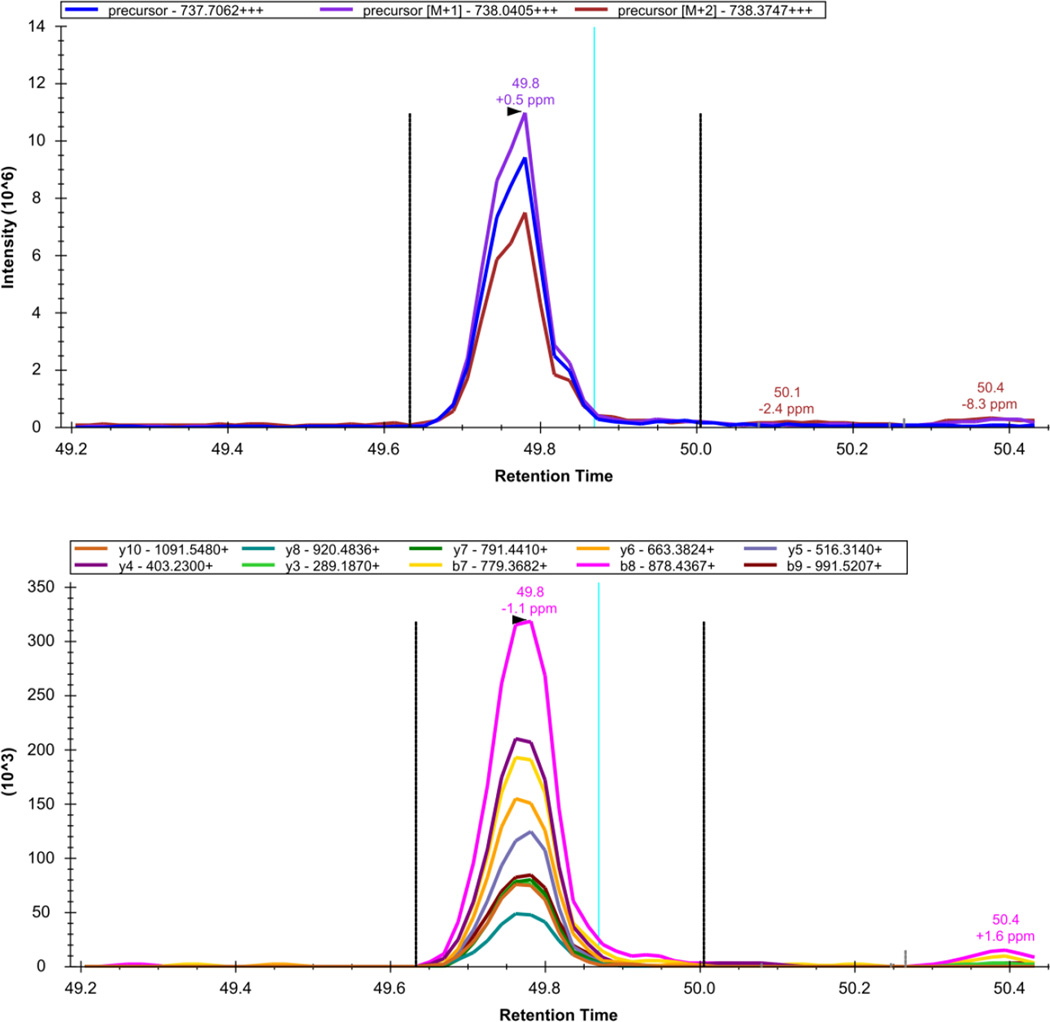
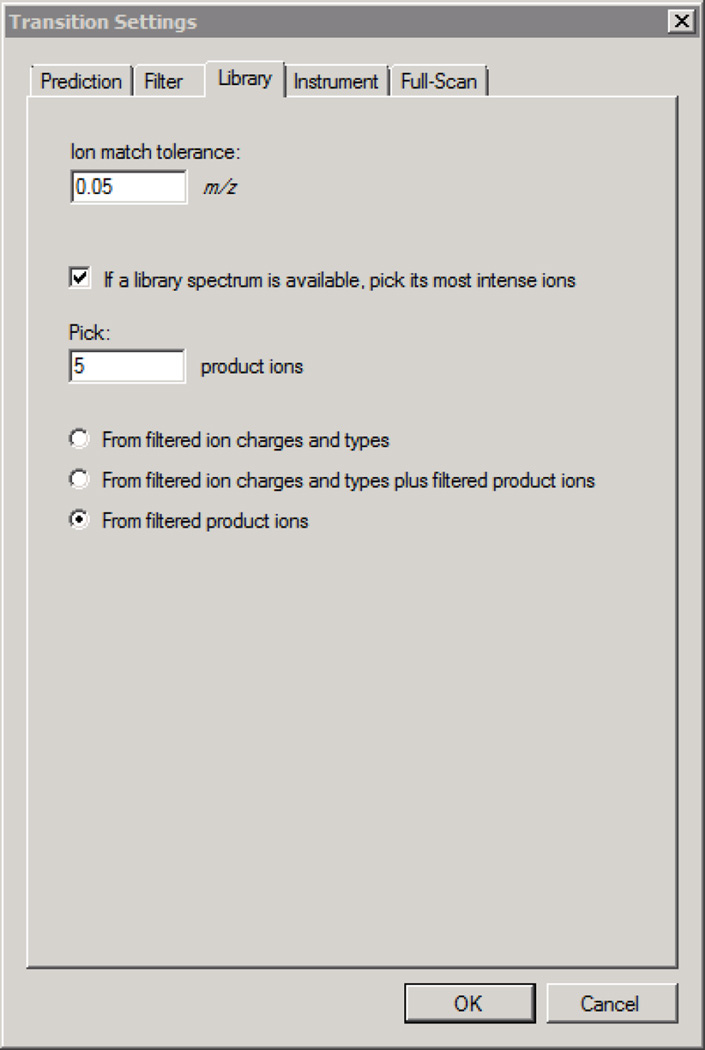
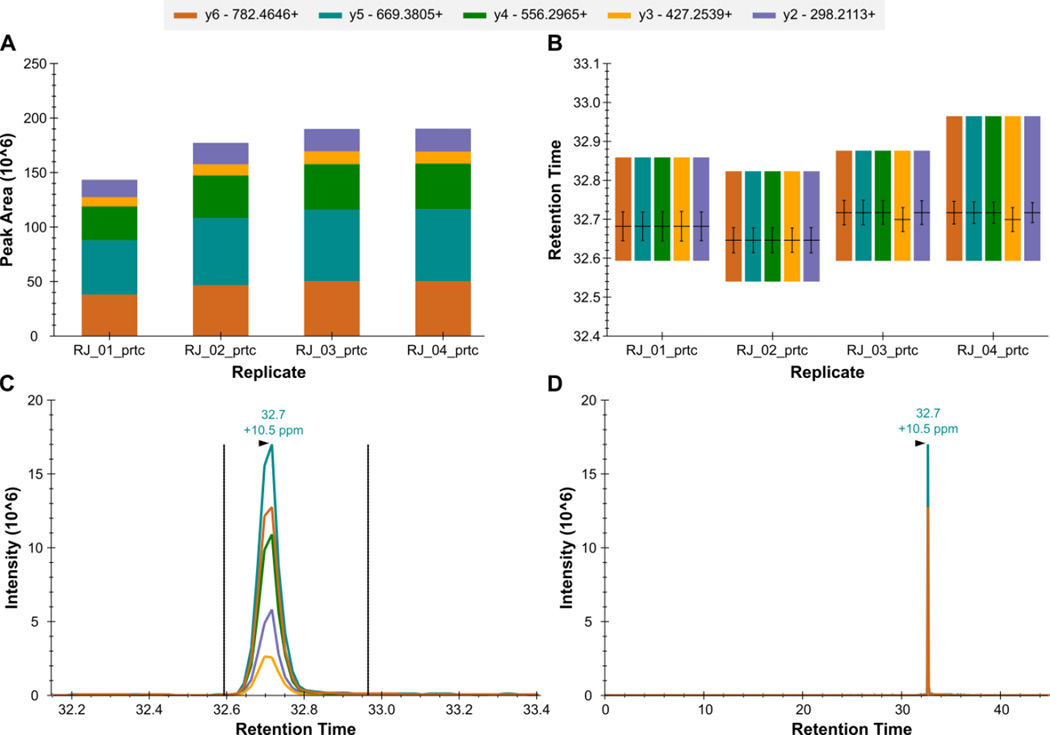
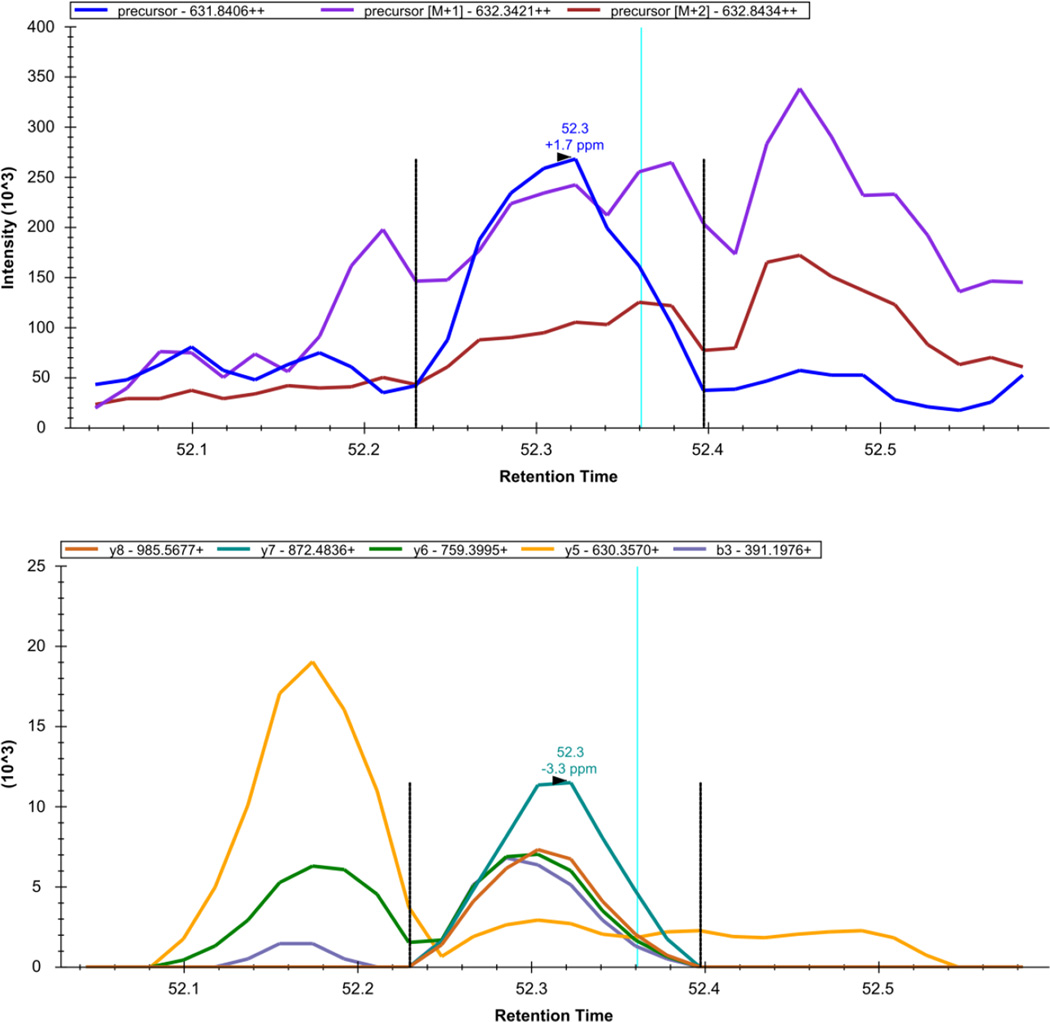
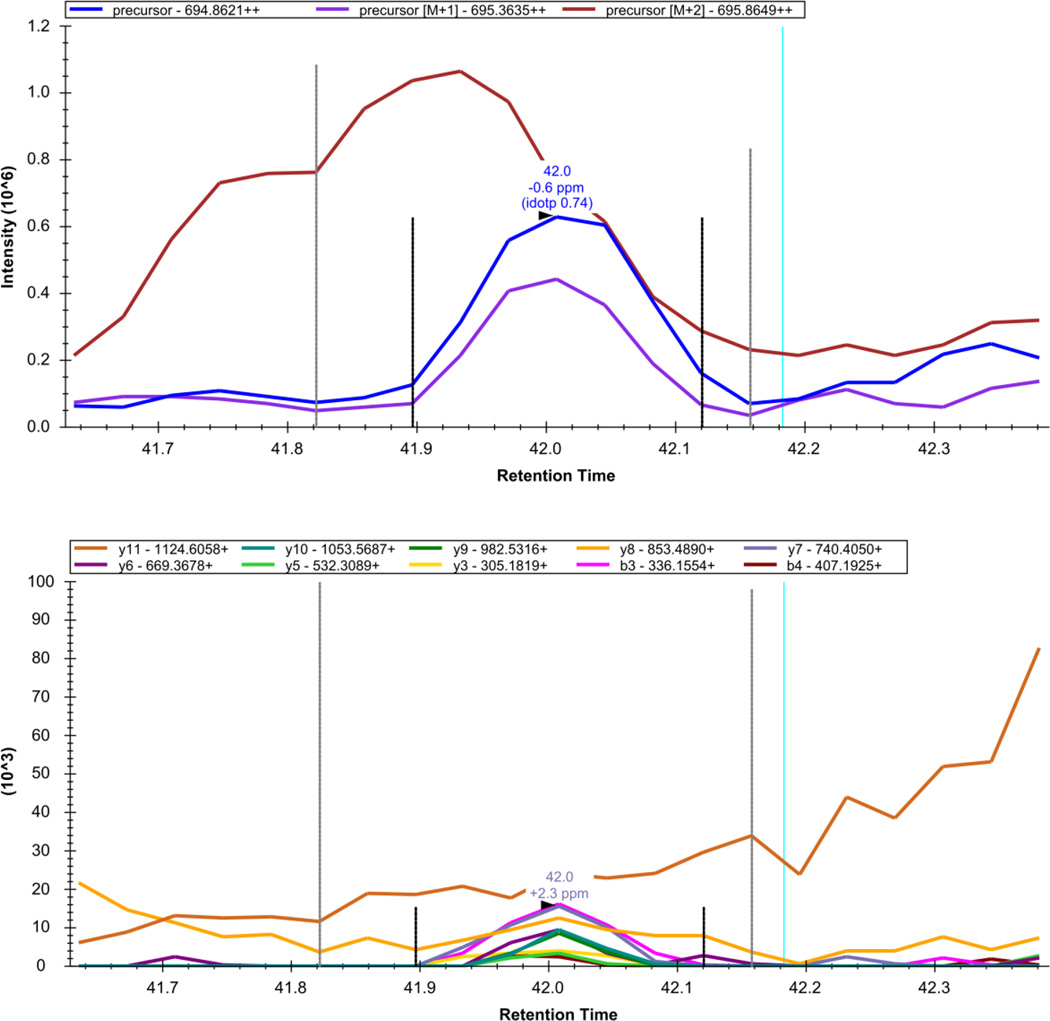
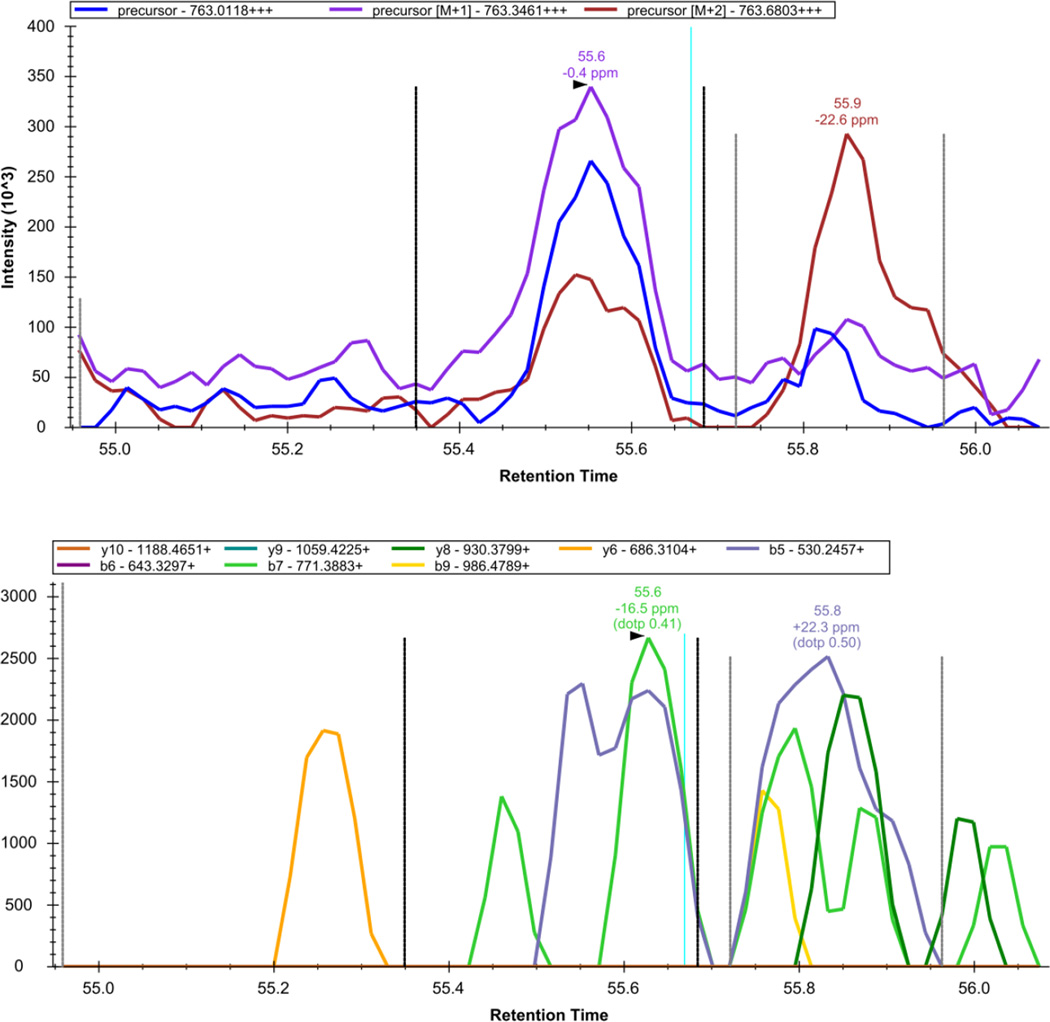
Here we describe the use of data-independent acquisition (DIA) on a Q-Exactive mass spectrometer for the detection and quantification of peptides in complex mixtures using the Skyline Targeted Proteomics Environment (freely available online at http://skyline.maccosslab.org). The systematic acquisition of mass spectrometry (MS) or tandem MS (MS/MS) spectra by DIA is in contrast to DDA, in which the acquired MS/MS spectra are only suitable for the identification of a stochastically sampled set of peptides. Similarly to selected reaction monitoring (SRM), peptides can be quantified from DIA data using targeted chromatogram extraction. Unlike SRM, data acquisition is not constrained to a predetermined set of target peptides. In this protocol, a spectral library is generated using data-dependent acquisition (DDA), and chromatograms are extracted from the DIA data for all peptides in the library. As in SRM, quantification using DIA data is based on the area under the curve of extracted MS/MS chromatograms. In addition, a quality control (QC) method suitable for DIA based on targeted MS/MS acquisition is detailed. Not including time spent acquiring data, and time for database searching, the procedure takes ∼1-2 h to complete. Typically, data acquisition requires roughly 1-4 h per sample, and a database search will take 0.5-2 h to complete.
Figures
References
-
- Purvine S, Eppel J-T, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3:847–850. - PubMed
-
- Venable JD, Dong M-Q, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods. 2004;1:39–45. - PubMed
-
- Law KP, Lim YP. Recent advances in mass spectrometry: data independent analysis and hyper reaction monitoring. Expert Rev. Proteomics. 2013;10:551–566. - PubMed
-
- Stahl-Zeng J, et al. High Sensitivity Detection of Plasma Proteins by Multiple Reaction Monitoring of N-Glycosites. Mol. Cell. Proteomics. 2007;6:1809–1817. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
