

Whole-genome sequencing for outbreak investigations of methicillin-resistant Staphylococcus aureus in the neonatal intensive care unit: time for routine practice?
- PMID: 25998499
- PMCID: PMC4507300
- DOI: 10.1017/ice.2015.73
Whole-genome sequencing for outbreak investigations of methicillin-resistant Staphylococcus aureus in the neonatal intensive care unit: time for routine practice?
Abstract
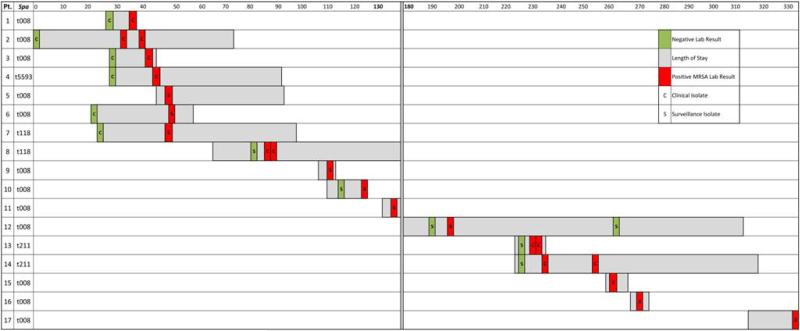
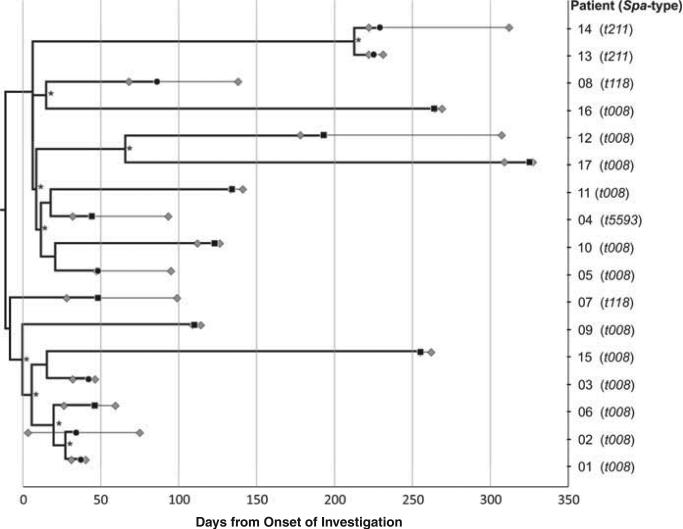
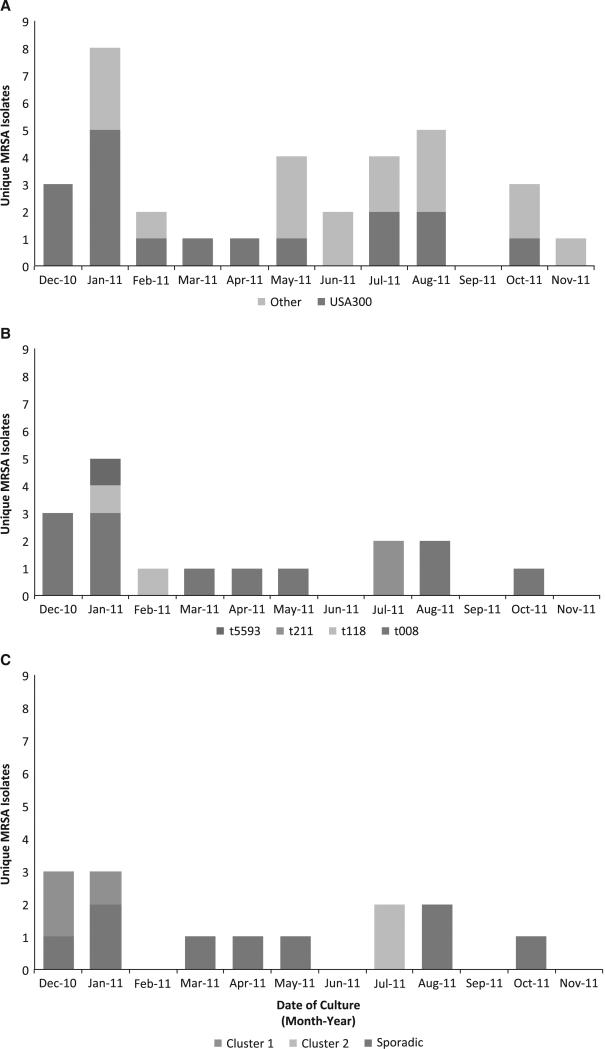
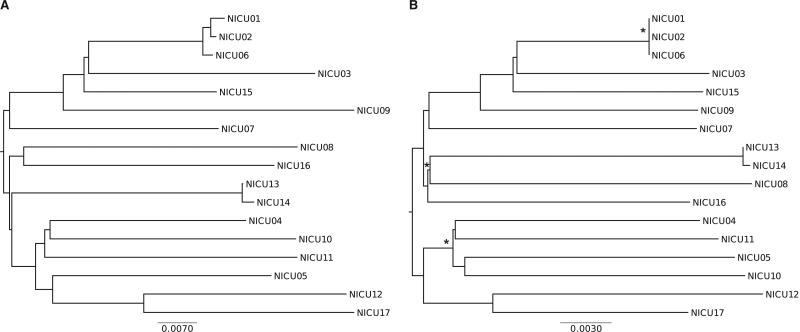
BACKGROUND Infants in the neonatal intensive care unit (NICU) are at increased risk for methicillin-resistant Staphylococcus aureus (MRSA) acquisition. Outbreaks may be difficult to identify due in part to limitations in current molecular genotyping available in clinical practice. Comparison of genome-wide single nucleotide polymorphisms (SNPs) may identify epidemiologically distinct isolates among a population sample that appears homogenous when evaluated using conventional typing methods. OBJECTIVE To investigate a putative MRSA outbreak in a NICU utilizing whole-genome sequencing and phylogenetic analysis to identify recent transmission events. DESIGN Clinical and surveillance specimens collected during clinical care and outbreak investigation. PATIENTS A total of 17 neonates hospitalized in a 43-bed level III NICU in northeastern Florida from December 2010 to October 2011 were included in this study. METHODS We assessed epidemiological data in conjunction with 4 typing methods: antibiograms, PFGE, spa types, and phylogenetic analysis of genome-wide SNPs. RESULTS Among the 17 type USA300 isolates, 4 different spa types were identified using pulsed-field gel electrophoresis. Phylogenetic analysis identified 5 infants as belonging to 2 clusters of epidemiologically linked cases and excluded 10 unlinked cases from putative transmission events. The availability of these results during the initial investigation would have improved infection control interventions. CONCLUSION Whole-genome sequencing and phylogenetic analysis are invaluable tools for epidemic investigation; they identify transmission events and exclude cases mistakenly implicated by traditional typing methods. When routinely applied to surveillance and investigation in the clinical setting, this approach may provide actionable intelligence for measured, appropriate, and effective interventions.
Figures
References
-
- Gerber SI, Jones RC, Scott MV, et al. Management of outbreaks of methicillin-resistant Staphylococcus aureus infection in the neonatal intensive care unit: a consensus statement. Infect Control Hosp Epidemiol. 2006;27:139–145. - PubMed
-
- Saiman L, Cronquist A, Wu F. An outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit. Infect Control Hosp Epidemiol. 2003;24:317–321. - PubMed
-
- Zervou FN, Zacharioudakis IM, Ziakas PD, Mylonakis E. MRSA colonization and risk of infection in the neonatal and pediatric ICU: a meta-analysis. Pediatrics. 2014;133:e1015–e1023. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
