

All-Atom Internal Coordinate Mechanics (ICM) Force Field for Hexopyranoses and Glycoproteins
- PMID: 25999804
- PMCID: PMC4431507
- DOI: 10.1021/ct501138c
All-Atom Internal Coordinate Mechanics (ICM) Force Field for Hexopyranoses and Glycoproteins
Abstract
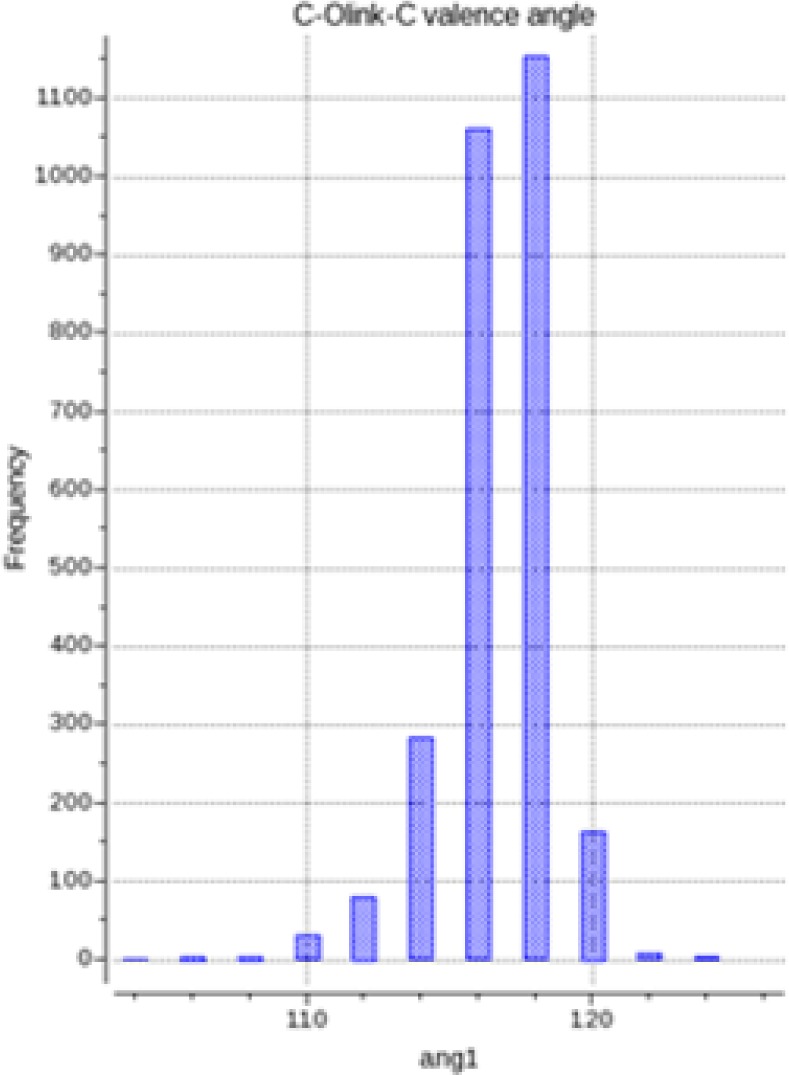
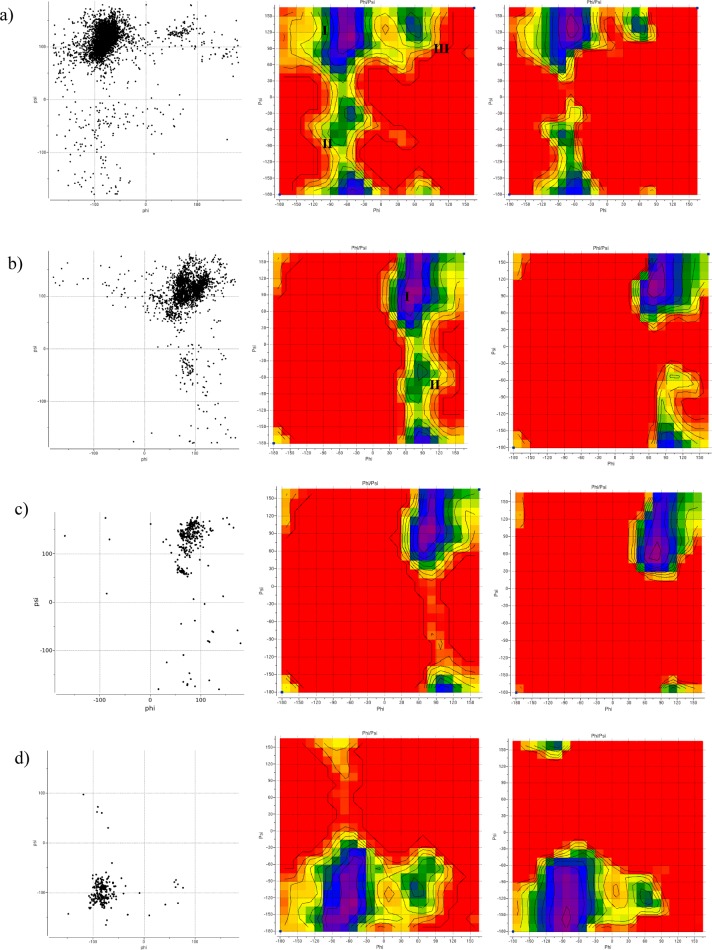
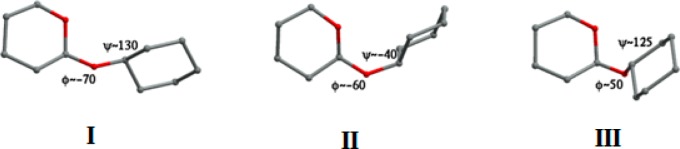
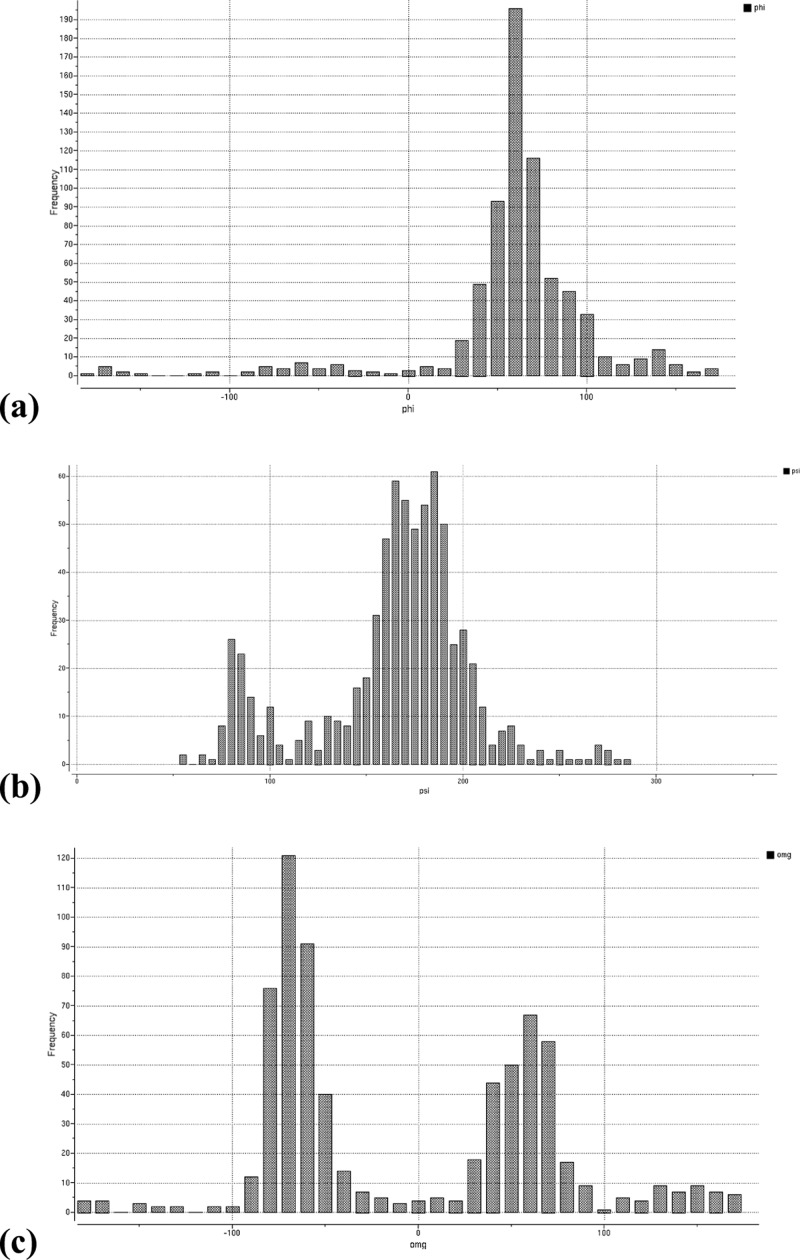
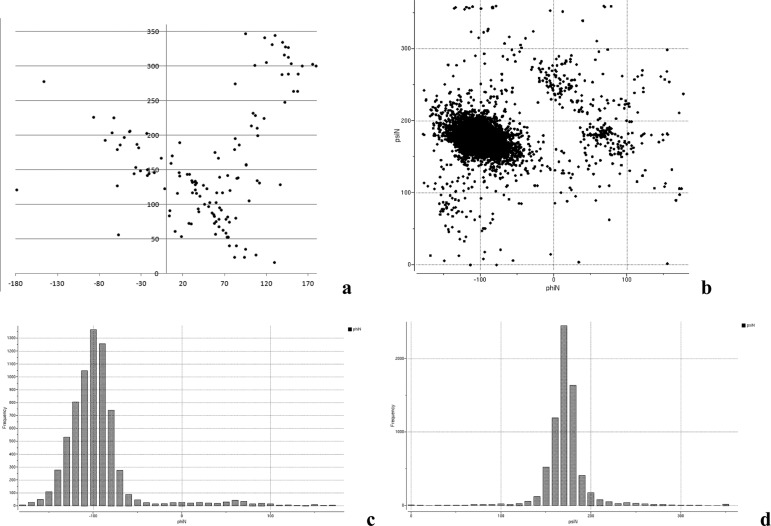
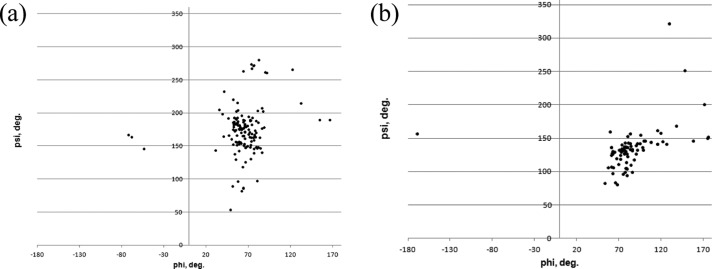
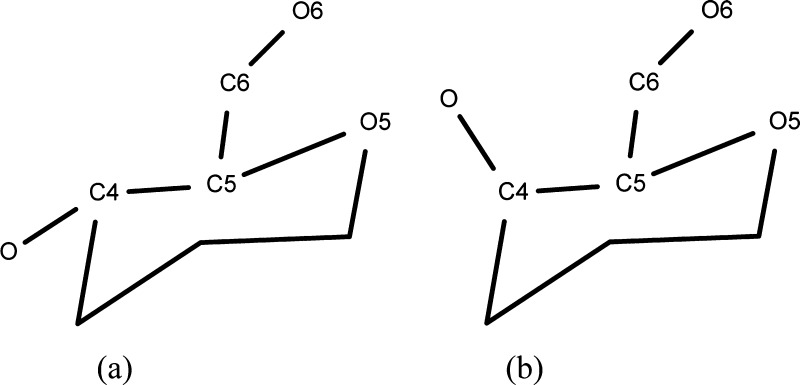
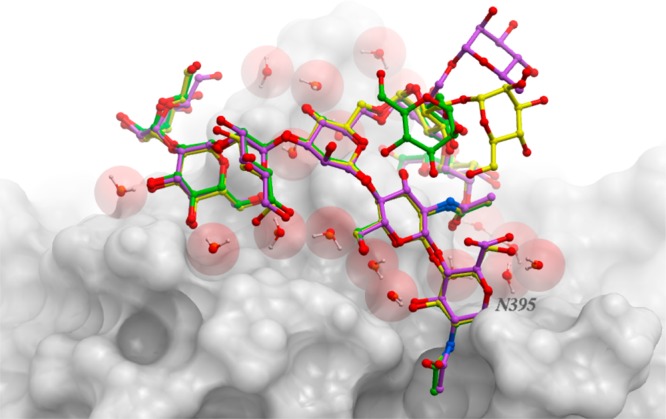
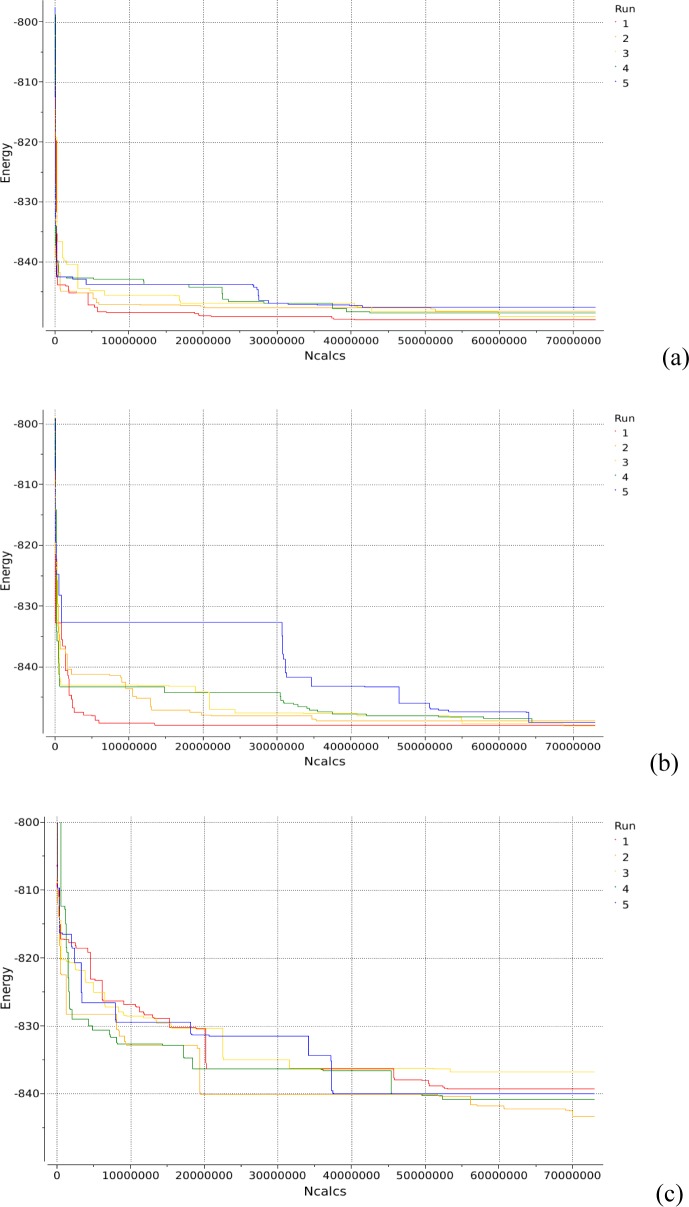
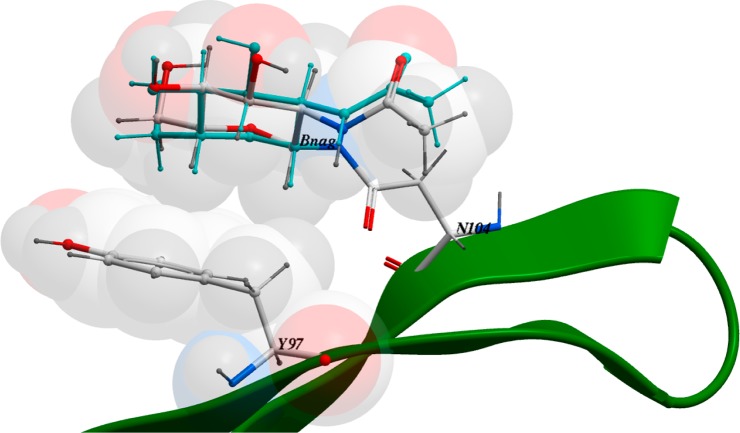
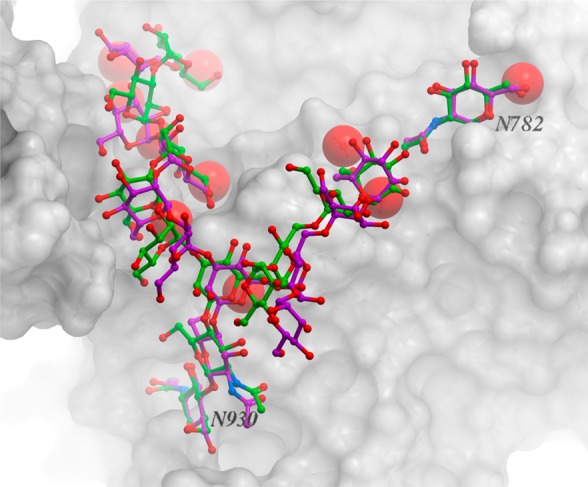
We present an extension of the all-atom internal-coordinate force field, ICMFF, that allows for simulation of heterogeneous systems including hexopyranose saccharides and glycan chains in addition to proteins. A library of standard glycan geometries containing α- and β-anomers of the most common hexapyranoses, i.e., d-galactose, d-glucose, d-mannose, d-xylose, l-fucose, N-acetylglucosamine, N-acetylgalactosamine, sialic, and glucuronic acids, is created based on the analysis of the saccharide structures reported in the Cambridge Structural Database. The new force field parameters include molecular electrostatic potential-derived partial atomic charges and the torsional parameters derived from quantum mechanical data for a collection of minimal molecular fragments and related molecules. The ϕ/ψ torsional parameters for different types of glycosidic linkages are developed using model compounds containing the key atoms in the full carbohydrates, i.e., glycosidic-linked tetrahydropyran-cyclohexane dimers. Target data for parameter optimization include two-dimensional energy surfaces corresponding to the ϕ/ψ glycosidic dihedral angles in the disaccharide analogues, as determined by quantum mechanical MP2/6-31G** single-point energies on HF/6-31G** optimized structures. To achieve better agreement with the observed geometries of glycosidic linkages, the bond angles at the O-linkage atoms are added to the internal variable set and the corresponding bond bending energy term is parametrized using quantum mechanical data. The resulting force field is validated on glycan chains of 1-12 residues from a set of high-resolution X-ray glycoprotein structures based on heavy atom root-mean-square deviations of the lowest-energy glycan conformations generated by the biased probability Monte Carlo (BPMC) molecular mechanics simulations from the native structures. The appropriate BPMC distributions for monosaccharide-monosaccharide and protein-glycan linkages are derived from the extensive analysis of conformational properties of glycoprotein structures reported in the Protein Data Bank. Use of the BPMC search leads to significant improvements in sampling efficiency for glycan simulations. Moreover, good agreement with the X-ray glycoprotein structures is achieved for all glycan chain lengths. Thus, average/median RMSDs are 0.81/0.68 Å for one-residue glycans and 1.32/1.47 Å for three-residue glycans. RMSD from the native structure for the lowest-energy conformation of the 12-residue glycan chain (PDB ID 3og2) is 1.53 Å. Additionally, results obtained for free short oligosaccharides using the new force field are in line with the available experimental data, i.e., the most populated conformations in solution are predicted to be the lowest energy ones. The newly developed parameters allow for the accurate modeling of linear and branched hexopyranose glycosides in heterogeneous systems.
Figures
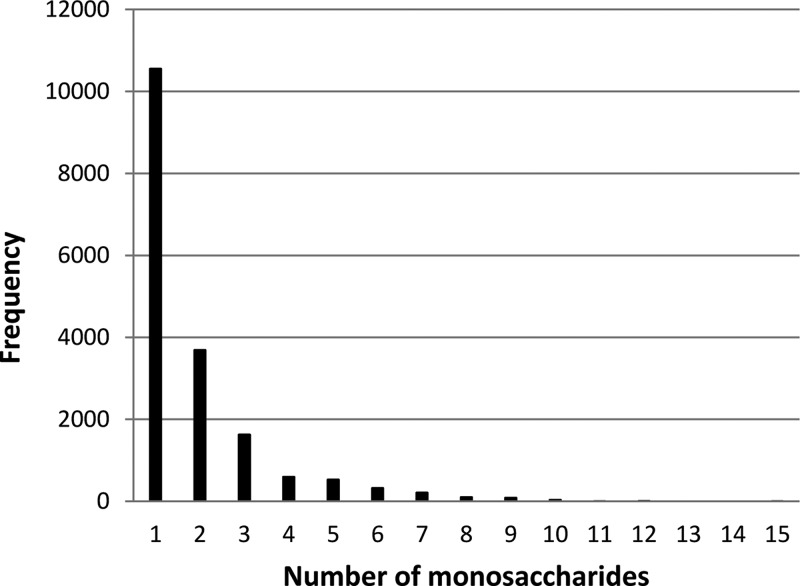
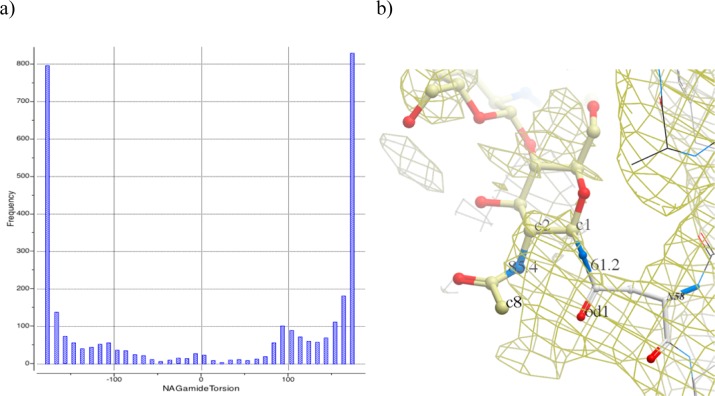
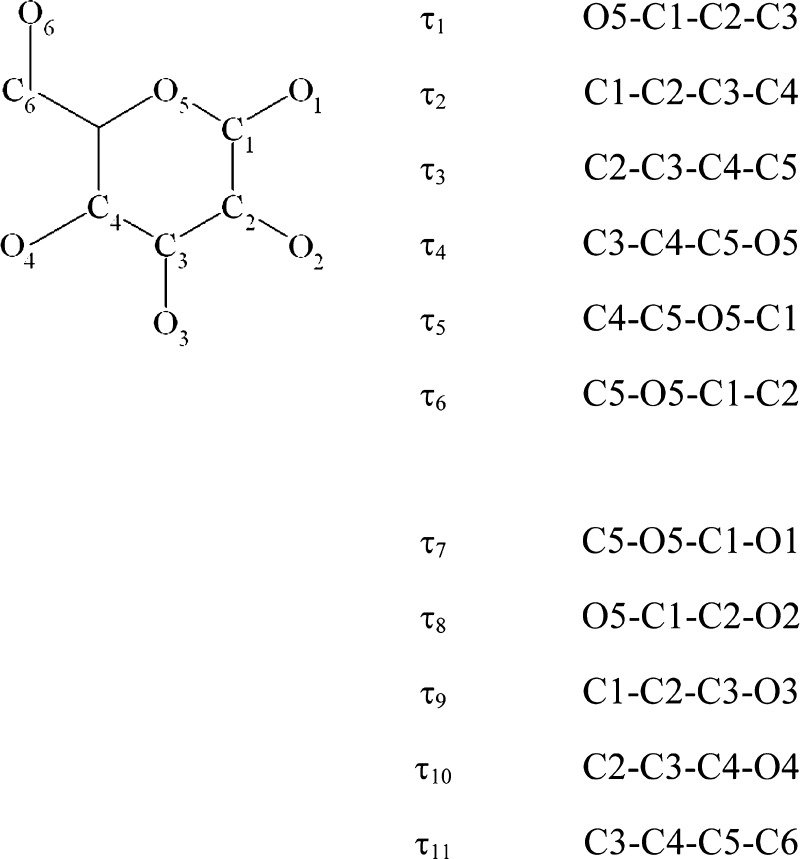
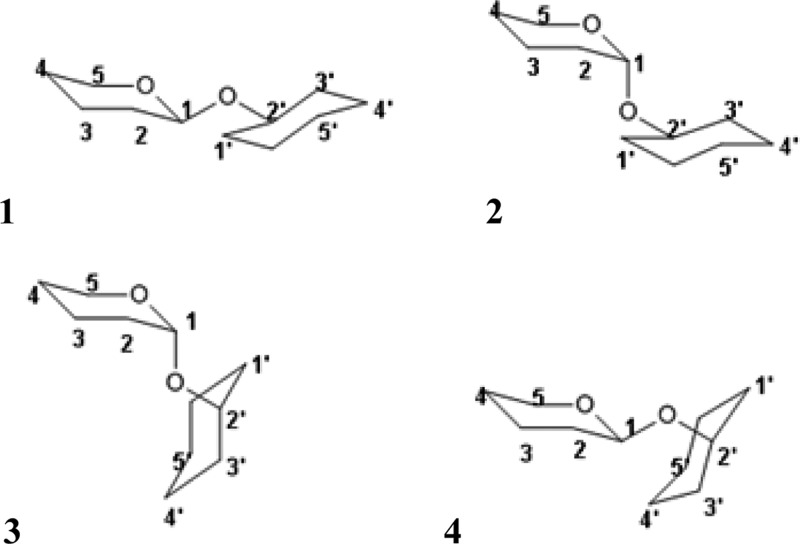
References
-
- Weinhold B.; Seidenfaden R.; Rockle I.; Muhlenhoff M.; Schertzinger F.; Conzelmann S.; Marth J. D.; Gerardy-Schahn R.; Hildebrandt H. J. Biol. Chem. 2005, 280, 42971. - PubMed
-
- Haltiwanger R. S.; Lowe J. B. Annu. Rev. Biochem. 2004, 73, 491. - PubMed
-
- Arnold J. N.; Wormald M. R.; Sim R. B.; Rudd P. M.; Dwek R. A. Annu. Rev. Immunol. 2007, 25, 21. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous