

Information flow and protein dynamics: the interplay between nuclear magnetic resonance spectroscopy and molecular dynamics simulations
- PMID: 25999971
- PMCID: PMC4419604
- DOI: 10.3389/fpls.2015.00306
Information flow and protein dynamics: the interplay between nuclear magnetic resonance spectroscopy and molecular dynamics simulations
Abstract
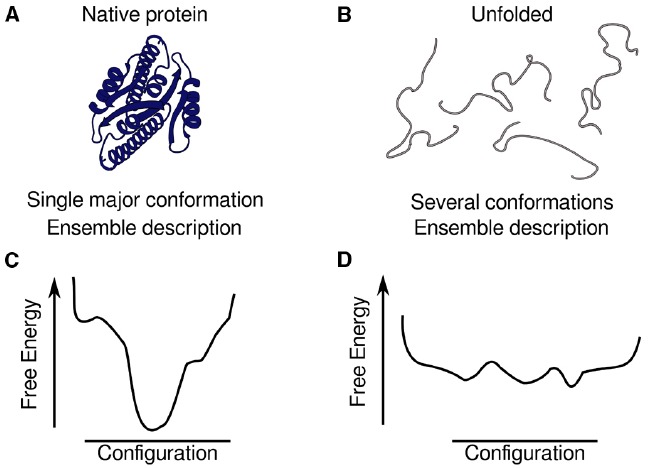
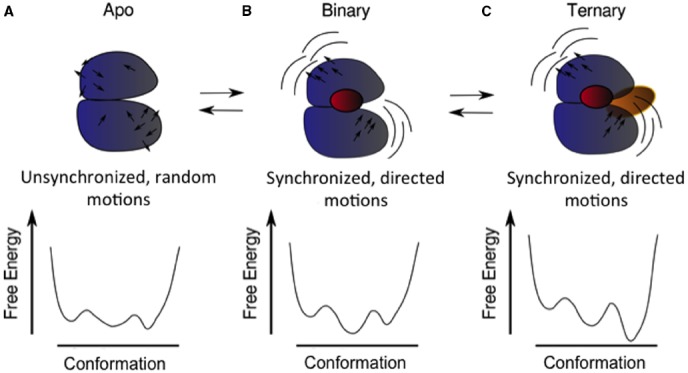
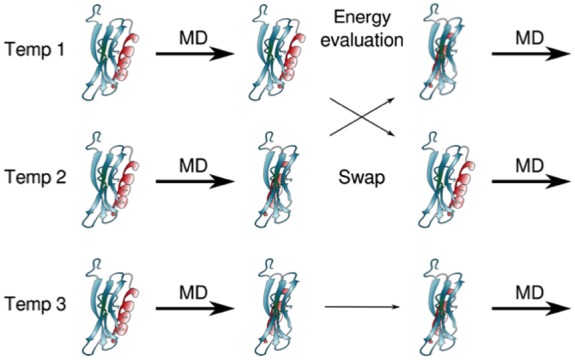
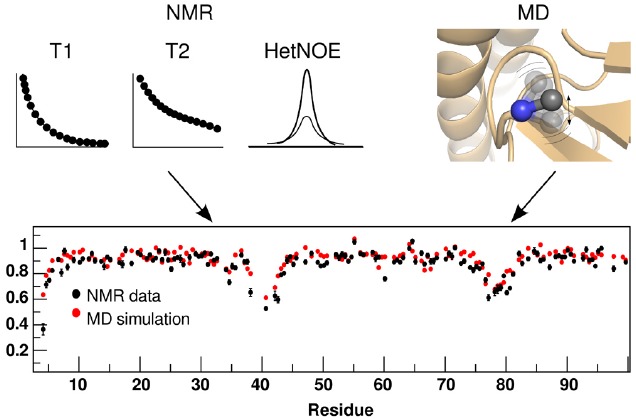
Proteins participate in information pathways in cells, both as links in the chain of signals, and as the ultimate effectors. Upon ligand binding, proteins undergo conformation and motion changes, which can be sensed by the following link in the chain of information. Nuclear magnetic resonance (NMR) spectroscopy and molecular dynamics (MD) simulations represent powerful tools for examining the time-dependent function of biological molecules. The recent advances in NMR and the availability of faster computers have opened the door to more detailed analyses of structure, dynamics, and interactions. Here we briefly describe the recent applications that allow NMR spectroscopy and MD simulations to offer unique insight into the basic motions that underlie information transfer within and between cells.
Keywords: aggregation; allosterism; binding; dynamics; folding intermediates.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources