

Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets
- PMID: 26000488
- PMCID: PMC4481139
- DOI: 10.1016/j.cell.2015.05.002
Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets
Abstract
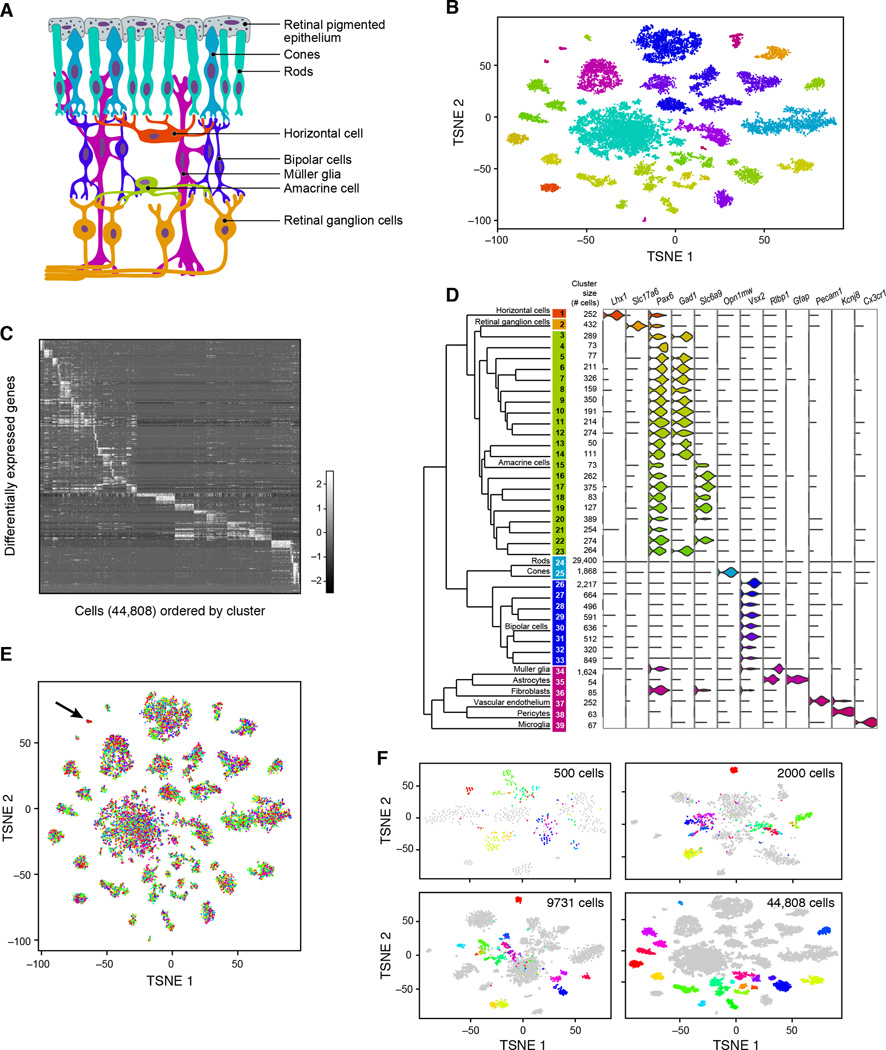
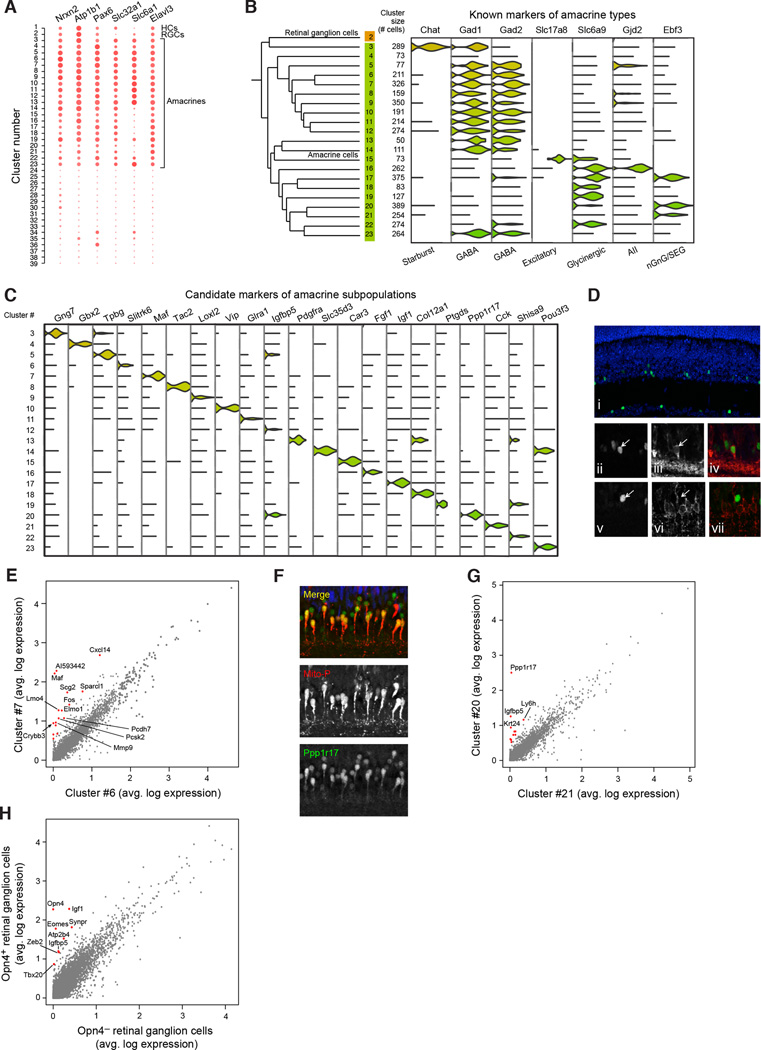
Cells, the basic units of biological structure and function, vary broadly in type and state. Single-cell genomics can characterize cell identity and function, but limitations of ease and scale have prevented its broad application. Here we describe Drop-seq, a strategy for quickly profiling thousands of individual cells by separating them into nanoliter-sized aqueous droplets, associating a different barcode with each cell's RNAs, and sequencing them all together. Drop-seq analyzes mRNA transcripts from thousands of individual cells simultaneously while remembering transcripts' cell of origin. We analyzed transcriptomes from 44,808 mouse retinal cells and identified 39 transcriptionally distinct cell populations, creating a molecular atlas of gene expression for known retinal cell classes and novel candidate cell subtypes. Drop-seq will accelerate biological discovery by enabling routine transcriptional profiling at single-cell resolution. VIDEO ABSTRACT.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
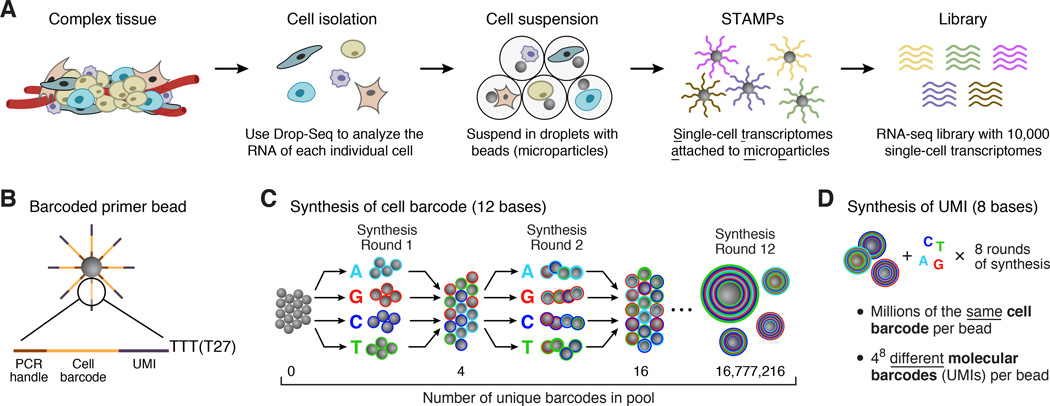
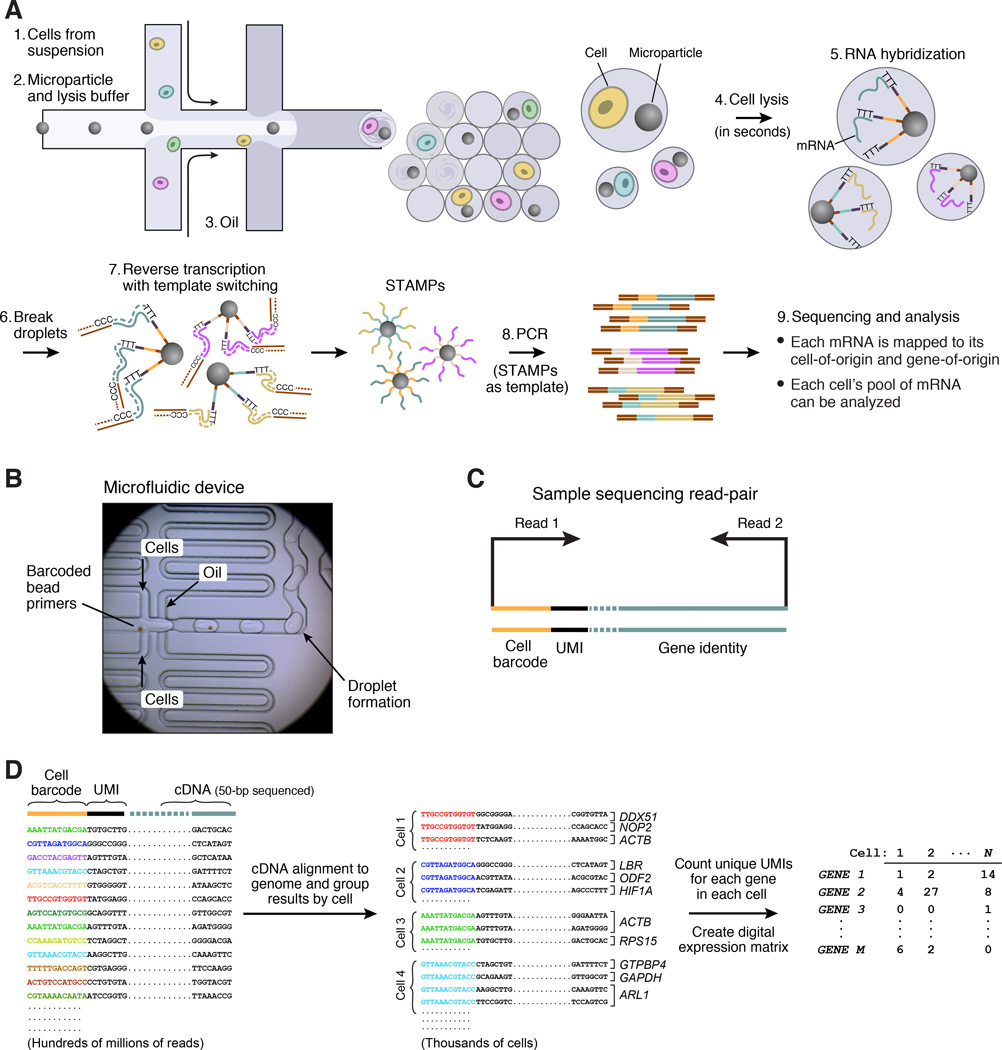
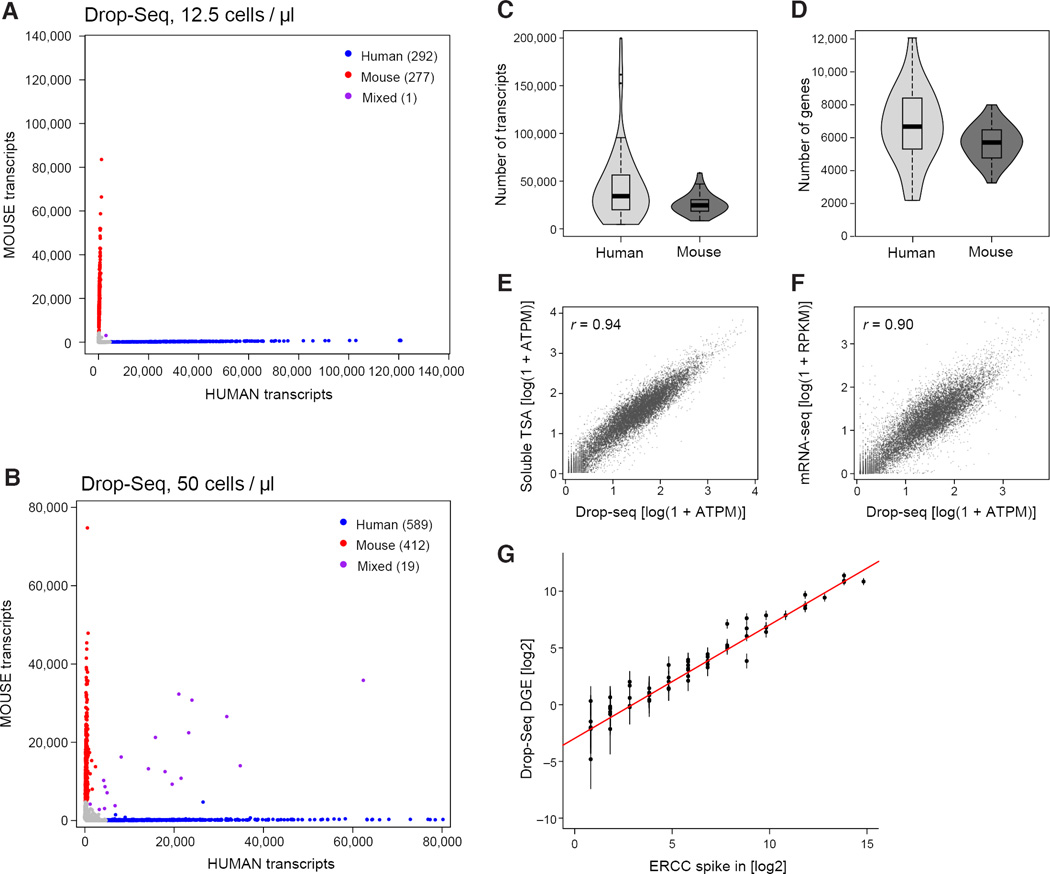
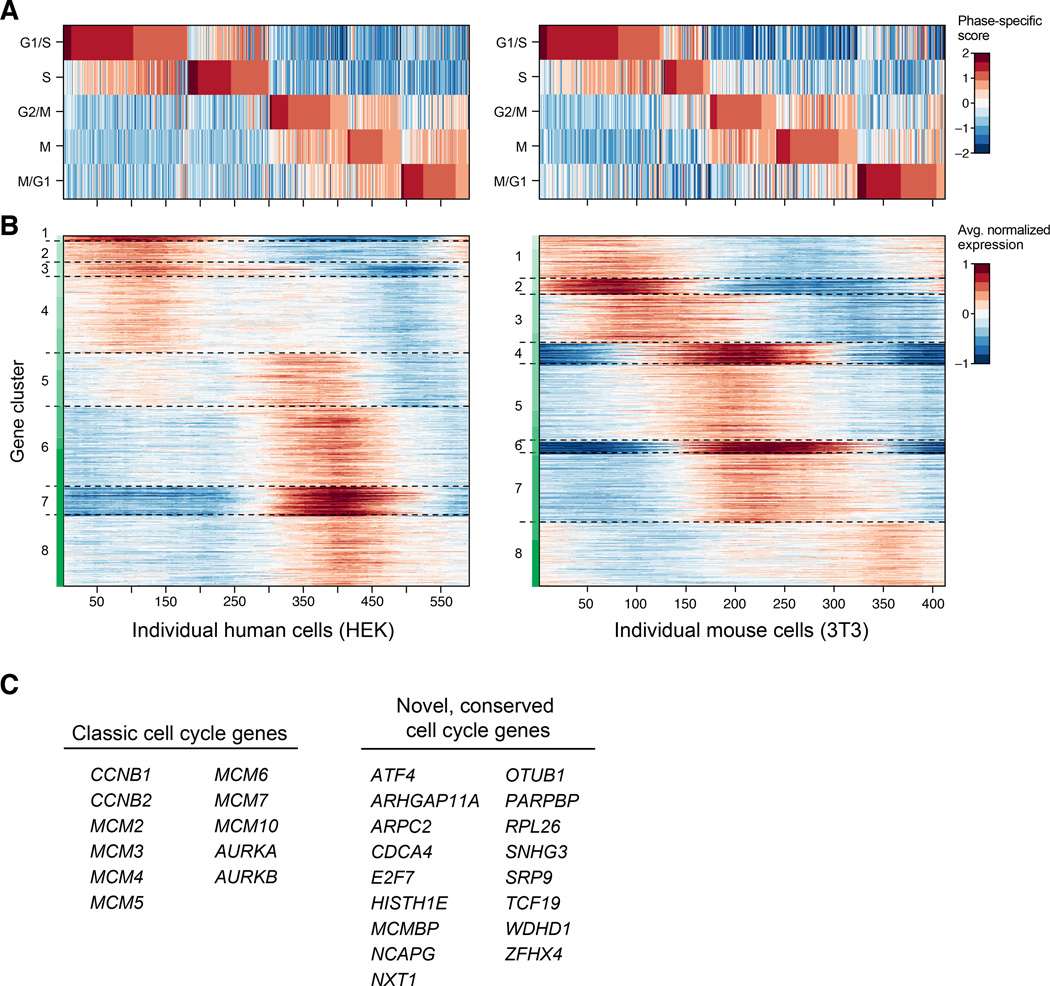
Comment in
-
Single-cell transcriptomics enters the age of mass production.Mol Cell. 2015 May 21;58(4):563-4. doi: 10.1016/j.molcel.2015.05.019. Mol Cell. 2015. PMID: 26000840
References
-
- Beer NR, Wheeler EK, Lee-Houghton L, Watkins N, Nasarabadi S, Hebert N, Leung P, Arnold DW, Bailey CG, Colston BW. On-chip single-copy real-time reverse-transcription PCR in isolated picoliter droplets. Analytical chemistry. 2008;80:1854–1858. - PubMed
-
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nature methods. 2013;10:1093–1095. - PubMed
-
- Britten RJ, Kohne DE. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science. 1968;161:529–540. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
