

Inexpensive multiplexed library preparation for megabase-sized genomes
- PMID: 26000737
- PMCID: PMC4441430
- DOI: 10.1371/journal.pone.0128036
Inexpensive multiplexed library preparation for megabase-sized genomes
Erratum in
-
Correction: Inexpensive Multiplexed Library Preparation for Megabase-Sized Genomes.PLoS One. 2015 Jun 18;10(6):e0131262. doi: 10.1371/journal.pone.0131262. eCollection 2015. PLoS One. 2015. PMID: 26086768 Free PMC article. No abstract available.
Abstract
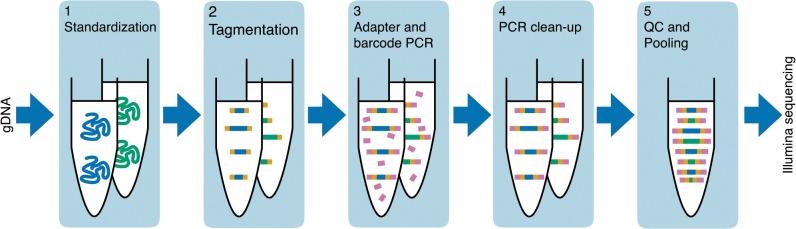
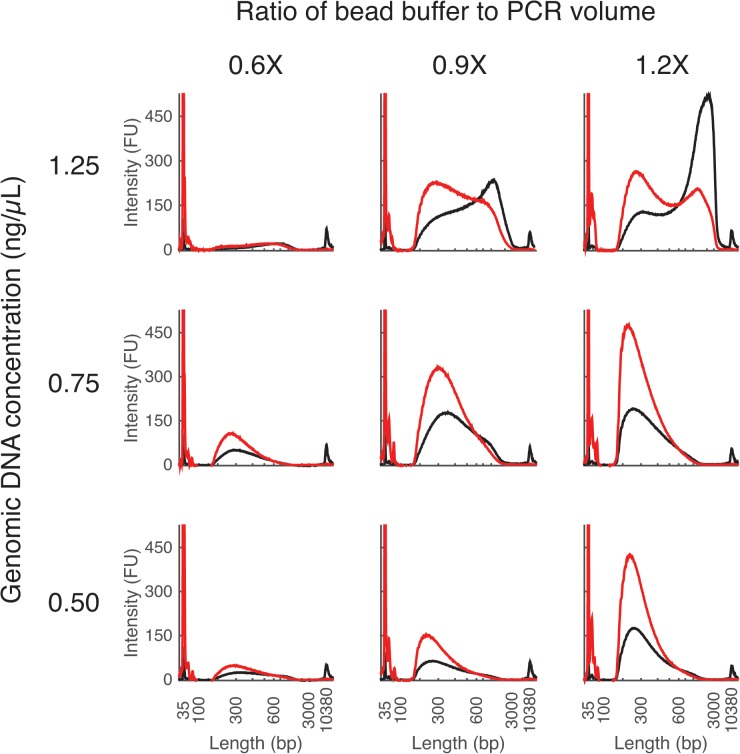
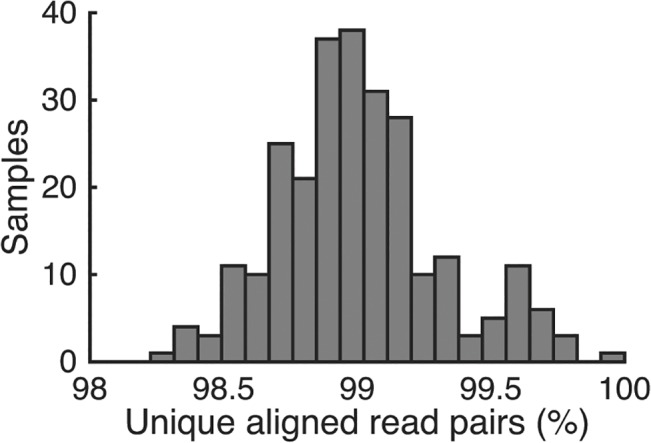
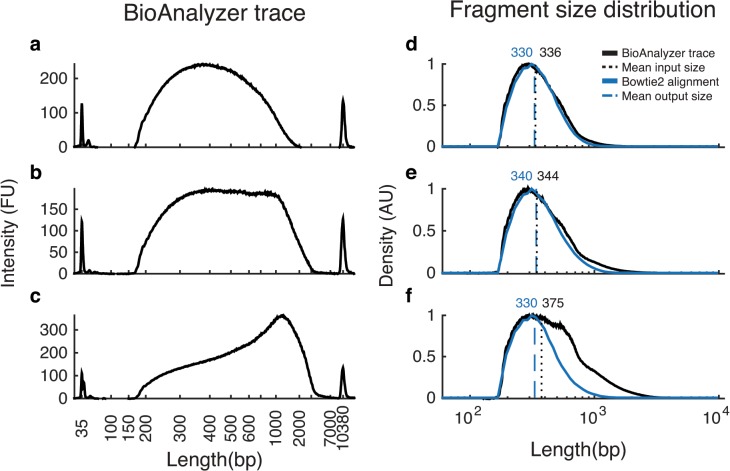
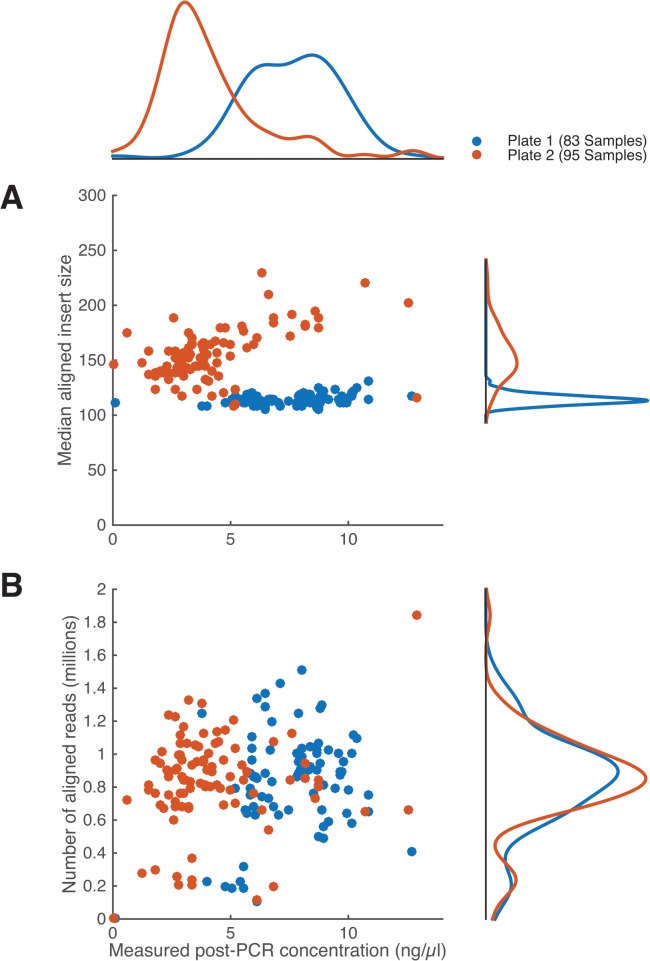
Whole-genome sequencing has become an indispensible tool of modern biology. However, the cost of sample preparation relative to the cost of sequencing remains high, especially for small genomes where the former is dominant. Here we present a protocol for rapid and inexpensive preparation of hundreds of multiplexed genomic libraries for Illumina sequencing. By carrying out the Nextera tagmentation reaction in small volumes, replacing costly reagents with cheaper equivalents, and omitting unnecessary steps, we achieve a cost of library preparation of $8 per sample, approximately 6 times cheaper than the standard Nextera XT protocol. Furthermore, our procedure takes less than 5 hours for 96 samples. Several hundred samples can then be pooled on the same HiSeq lane via custom barcodes. Our method will be useful for re-sequencing of microbial or viral genomes, including those from evolution experiments, genetic screens, and environmental samples, as well as for other sequencing applications including large amplicon, open chromosome, artificial chromosomes, and RNA sequencing.
Conflict of interest statement
Figures
References
-
- Mardis ER (2008) Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
