

TP53 Silencing Bypasses Growth Arrest of BRAFV600E-Induced Lung Tumor Cells in a Two-Switch Model of Lung Tumorigenesis
- PMID: 26001956
- PMCID: PMC4526430
- DOI: 10.1158/0008-5472.CAN-14-3701
TP53 Silencing Bypasses Growth Arrest of BRAFV600E-Induced Lung Tumor Cells in a Two-Switch Model of Lung Tumorigenesis
Abstract
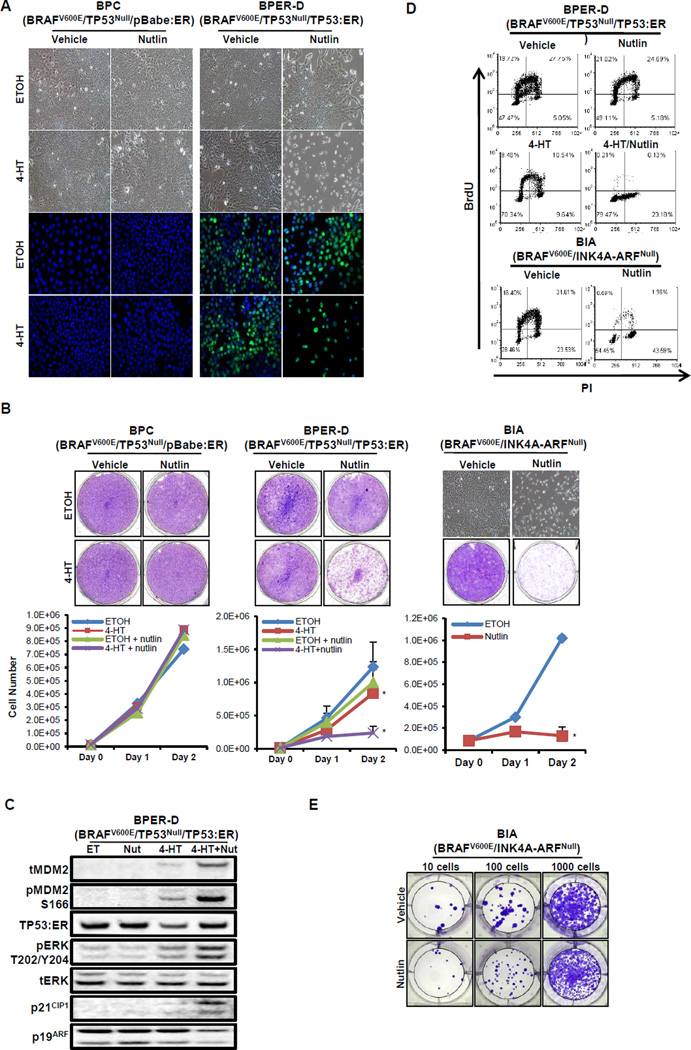
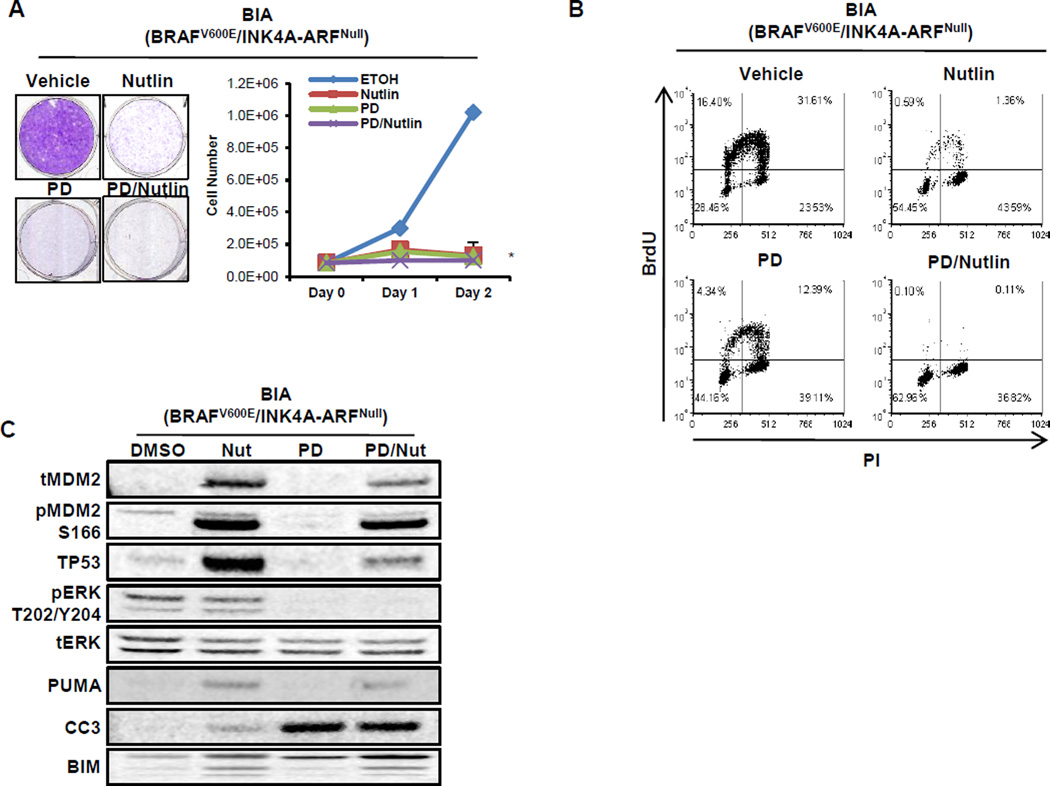
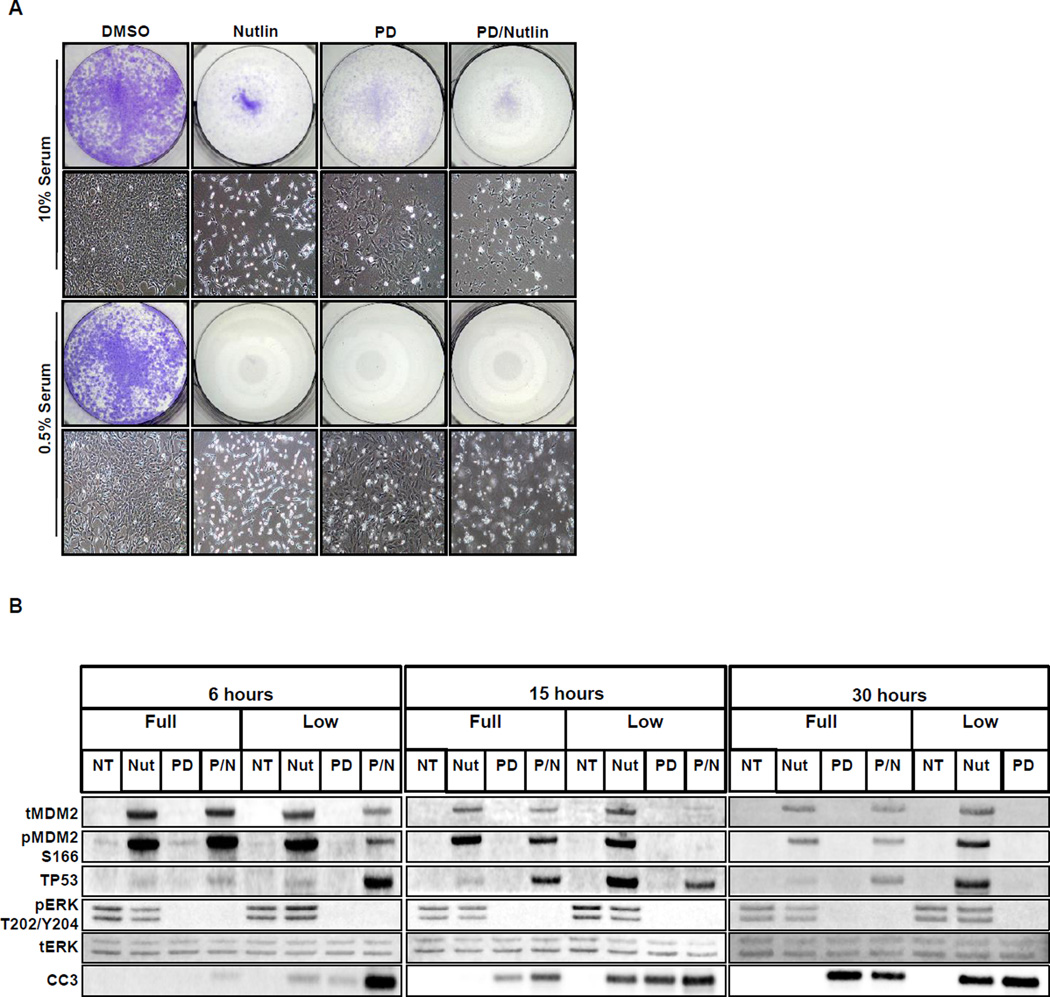
Lung carcinogenesis is a multistep process in which normal lung epithelial cells are converted to cancer cells through the sequential acquisition of multiple genetic or epigenetic events. Despite the utility of current genetically engineered mouse (GEM) models of lung cancer, most do not allow temporal dissociation of the cardinal events involved in lung tumor initiation and cancer progression. Here we describe a novel two-switch GEM model for BRAF(V600E)-induced lung carcinogenesis allowing temporal dissociation of these processes. In mice carrying a Flp recombinase-activated allele of Braf (Braf(FA)) in conjunction with Cre-regulated alleles of Trp53, Cdkn2a, or c-MYC, we demonstrate that secondary genetic events can promote bypass of the senescence-like proliferative arrest displayed by BRAF(V600E)-induced lung adenomas, leading to malignant progression. Moreover, restoring or activating TP53 in cultured BRAF(V600E)/TP53(Null) or BRAF(V600E)/INK4A-ARF(Null) lung cancer cells triggered a G1 cell-cycle arrest regardless of p19(ARF) status. Perhaps surprisingly, neither senescence nor apoptosis was observed upon TP53 restoration. Our results establish a central function for the TP53 pathway in restricting lung cancer development, highlighting the mechanisms that limit malignant progression of BRAF(V600E)-initiated tumors.
©2015 American Association for Cancer Research.
Figures
Similar articles
-
MEK1/2 inhibition elicits regression of autochthonous lung tumors induced by KRASG12D or BRAFV600E.Cancer Res. 2012 Jun 15;72(12):3048-59. doi: 10.1158/0008-5472.CAN-11-3649. Epub 2012 Apr 17. Cancer Res. 2012. PMID: 22511580 Free PMC article.
-
p53 loss does not permit escape from BrafV600E-induced senescence in a mouse model of lung cancer.Oncogene. 2017 Nov 9;36(45):6325-6335. doi: 10.1038/onc.2017.235. Epub 2017 Jul 24. Oncogene. 2017. PMID: 28745322
-
A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors.Genes Dev. 2007 Feb 15;21(4):379-84. doi: 10.1101/gad.1516407. Epub 2007 Feb 13. Genes Dev. 2007. PMID: 17299132 Free PMC article.
-
Mouse Models as a Tool for Understanding Progression in BrafV600E-Driven Thyroid Cancers.Endocrinol Metab (Seoul). 2019 Mar;34(1):11-22. doi: 10.3803/EnM.2019.34.1.11. Epub 2019 Feb 15. Endocrinol Metab (Seoul). 2019. PMID: 30784243 Free PMC article. Review.
-
Mouse models for BRAF-induced cancers.Biochem Soc Trans. 2007 Nov;35(Pt 5):1329-33. doi: 10.1042/BST0351329. Biochem Soc Trans. 2007. PMID: 17956344 Free PMC article. Review.
Cited by
-
TFAP2C promotes lung tumorigenesis and aggressiveness through miR-183- and miR-33a-mediated cell cycle regulation.Oncogene. 2017 Mar;36(11):1585-1596. doi: 10.1038/onc.2016.328. Epub 2016 Sep 5. Oncogene. 2017. PMID: 27593936
-
Tissue specificity of oncogenic BRAF targeted to lung and thyroid through a shared lineage factor.iScience. 2023 Jun 8;26(7):107071. doi: 10.1016/j.isci.2023.107071. eCollection 2023 Jul 21. iScience. 2023. PMID: 37534159 Free PMC article.
-
FoxA1 and FoxA2 control growth and cellular identity in NKX2-1-positive lung adenocarcinoma.Dev Cell. 2022 Aug 8;57(15):1866-1882.e10. doi: 10.1016/j.devcel.2022.06.017. Epub 2022 Jul 13. Dev Cell. 2022. PMID: 35835117 Free PMC article.
-
A Transgenic MMTV-Flippase Mouse Line for Molecular Engineering in Mammary Gland and Breast Cancer Mouse Models.J Mammary Gland Biol Neoplasia. 2019 Mar;24(1):39-45. doi: 10.1007/s10911-018-9412-4. Epub 2018 Sep 12. J Mammary Gland Biol Neoplasia. 2019. PMID: 30209717
-
Early differential responses elicited by BRAFV600E in adult mouse models.Cell Death Dis. 2022 Feb 10;13(2):142. doi: 10.1038/s41419-022-04597-z. Cell Death Dis. 2022. PMID: 35145078 Free PMC article.
References
-
- Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the Surveillance, Epidemiology, and End Results (SEER) Program. Oncologist. 2007;12(1):20–37. - PubMed
-
- Heist RS, Engelman JA. SnapShot: non-small cell lung cancer. Cancer Cell. 2012;21(3):448 e2. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
