

Global transcription network incorporating distal regulator binding reveals selective cooperation of cancer drivers and risk genes
- PMID: 26001967
- PMCID: PMC4499150
- DOI: 10.1093/nar/gkv532
Global transcription network incorporating distal regulator binding reveals selective cooperation of cancer drivers and risk genes
Abstract
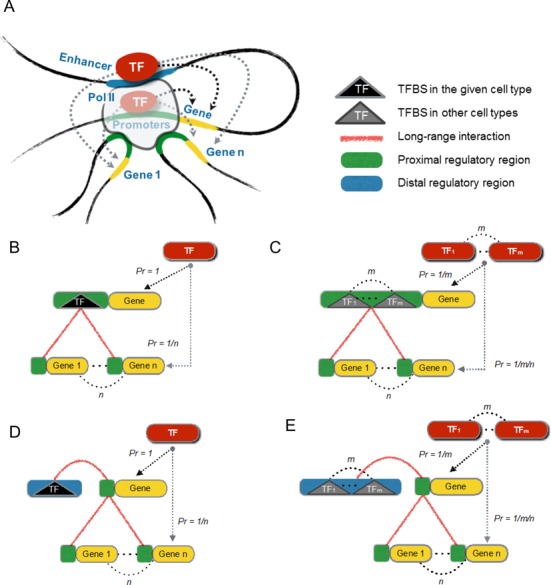
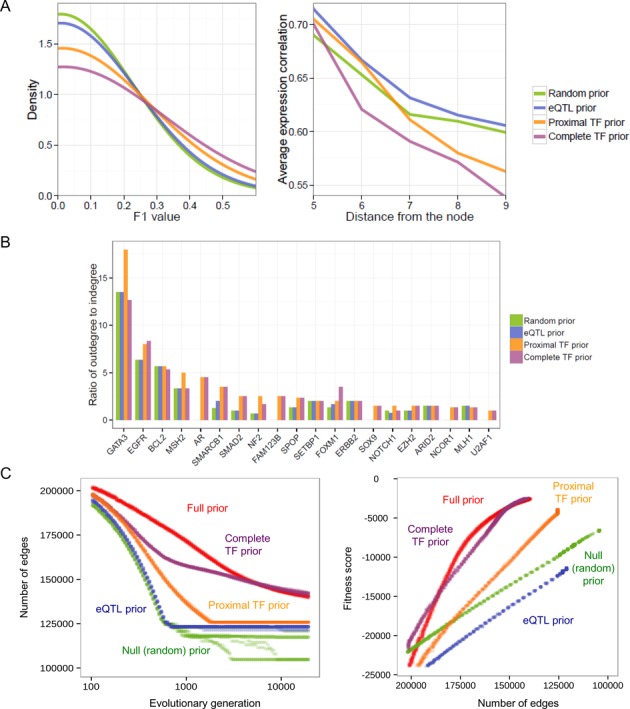
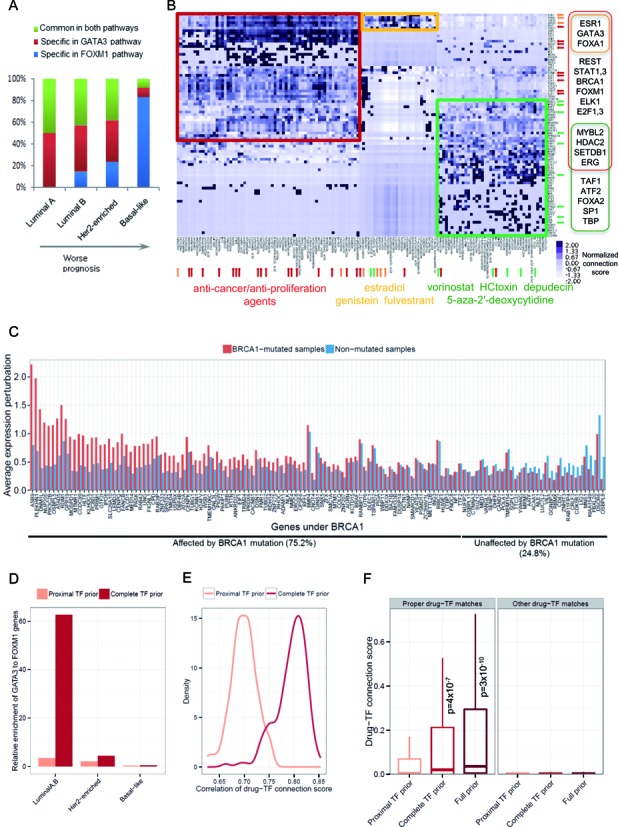
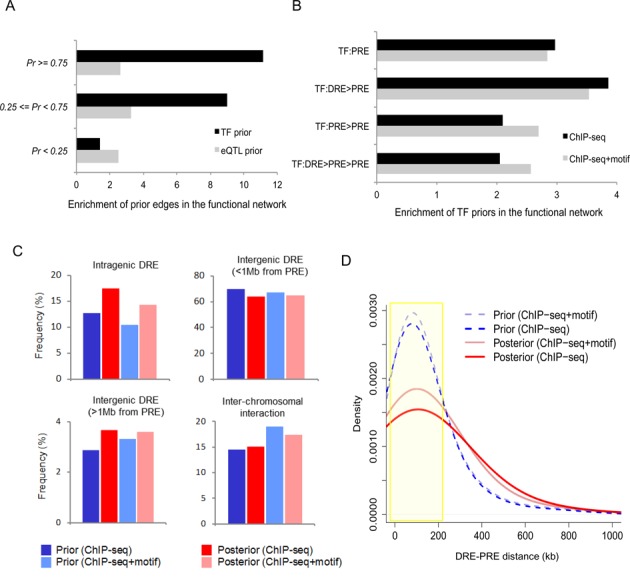
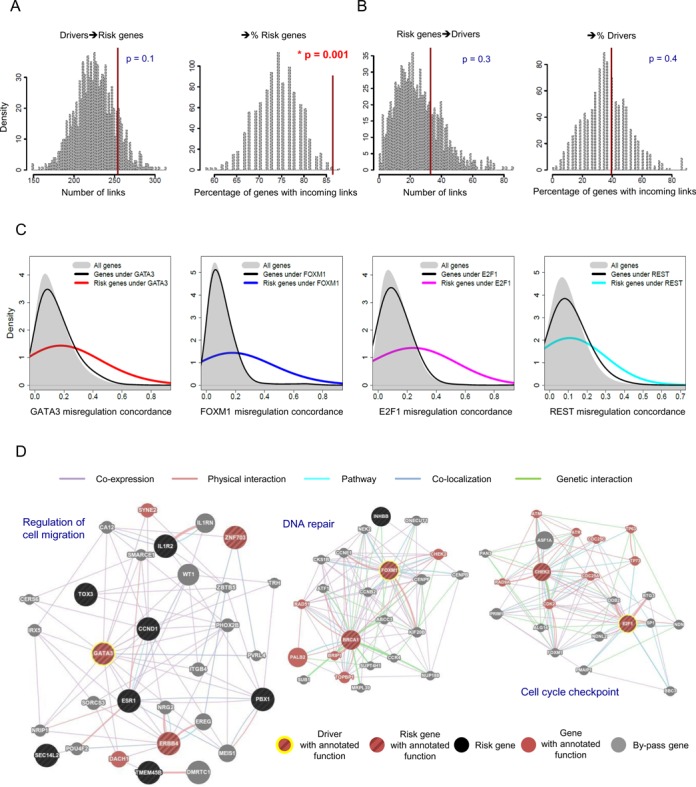
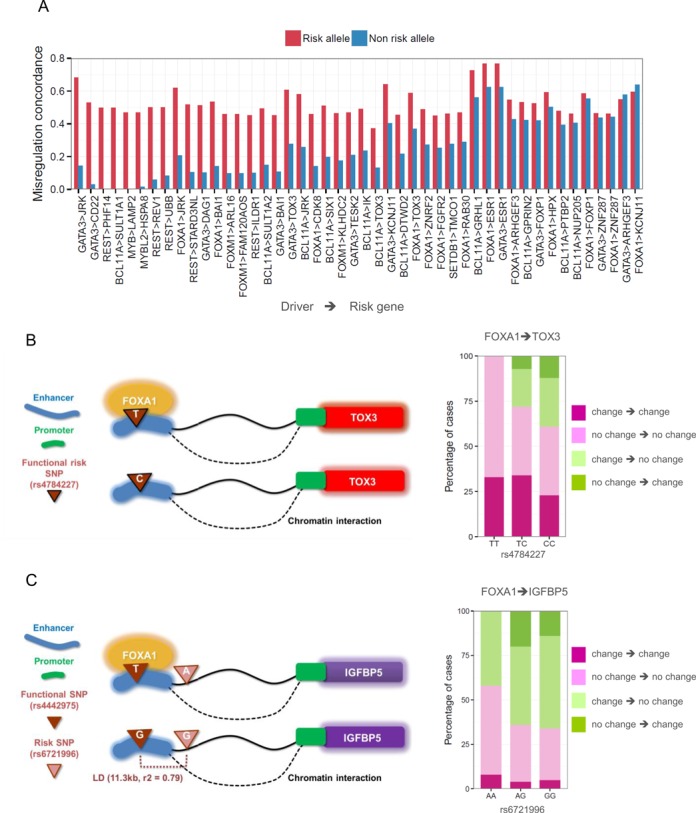
Global network modeling of distal regulatory interactions is essential in understanding the overall architecture of gene expression programs. Here, we developed a Bayesian probabilistic model and computational method for global causal network construction with breast cancer as a model. Whereas physical regulator binding was well supported by gene expression causality in general, distal elements in intragenic regions or loci distant from the target gene exhibited particularly strong functional effects. Modeling the action of long-range enhancers was critical in recovering true biological interactions with increased coverage and specificity overall and unraveling regulatory complexity underlying tumor subclasses and drug responses in particular. Transcriptional cancer drivers and risk genes were discovered based on the network analysis of somatic and genetic cancer-related DNA variants. Notably, we observed that the risk genes were functionally downstream of the cancer drivers and were selectively susceptible to network perturbation by tumorigenic changes in their upstream drivers. Furthermore, cancer risk alleles tended to increase the susceptibility of the transcription of their associated genes. These findings suggest that transcriptional cancer drivers selectively induce a combinatorial misregulation of downstream risk genes, and that genetic risk factors, mostly residing in distal regulatory regions, increase transcriptional susceptibility to upstream cancer-driving somatic changes.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Network perturbation by recurrent regulatory variants in cancer.PLoS Comput Biol. 2017 Mar 23;13(3):e1005449. doi: 10.1371/journal.pcbi.1005449. eCollection 2017 Mar. PLoS Comput Biol. 2017. PMID: 28333928 Free PMC article.
-
Inferring regulatory element landscapes and transcription factor networks from cancer methylomes.Genome Biol. 2015 May 21;16(1):105. doi: 10.1186/s13059-015-0668-3. Genome Biol. 2015. PMID: 25994056 Free PMC article.
-
The DtxR protein acting as dual transcriptional regulator directs a global regulatory network involved in iron metabolism of Corynebacterium glutamicum.BMC Genomics. 2006 Feb 9;7:21. doi: 10.1186/1471-2164-7-21. BMC Genomics. 2006. PMID: 16469103 Free PMC article.
-
Long-range transcriptional regulation of breast cancer genes.Genes Chromosomes Cancer. 2013 Feb;52(2):113-25. doi: 10.1002/gcc.22020. Epub 2012 Oct 18. Genes Chromosomes Cancer. 2013. PMID: 23077082 Review.
-
Advances in analysis of transcriptional regulatory networks.Wiley Interdiscip Rev Syst Biol Med. 2011 Jan-Feb;3(1):21-35. doi: 10.1002/wsbm.105. Wiley Interdiscip Rev Syst Biol Med. 2011. PMID: 21069662 Review.
Cited by
-
Predicting the recurrence of noncoding regulatory mutations in cancer.BMC Bioinformatics. 2016 Dec 3;17(1):492. doi: 10.1186/s12859-016-1385-y. BMC Bioinformatics. 2016. PMID: 27912731 Free PMC article.
-
Network perturbation by recurrent regulatory variants in cancer.PLoS Comput Biol. 2017 Mar 23;13(3):e1005449. doi: 10.1371/journal.pcbi.1005449. eCollection 2017 Mar. PLoS Comput Biol. 2017. PMID: 28333928 Free PMC article.
-
Computational inference of cancer-specific vulnerabilities in clinical samples.Genome Biol. 2020 Jun 29;21(1):155. doi: 10.1186/s13059-020-02077-1. Genome Biol. 2020. PMID: 32600395 Free PMC article.
-
NFP: An R Package for Characterizing and Comparing of Annotated Biological Networks.Biomed Res Int. 2017;2017:7457131. doi: 10.1155/2017/7457131. Epub 2017 Feb 9. Biomed Res Int. 2017. PMID: 28280740 Free PMC article.
-
How to interpret and integrate multi-omics data at systems level.Anim Cells Syst (Seoul). 2020 Jan 30;24(1):1-7. doi: 10.1080/19768354.2020.1721321. eCollection 2020. Anim Cells Syst (Seoul). 2020. PMID: 32158610 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
