

Macrophage Mitochondrial Energy Status Regulates Cholesterol Efflux and Is Enhanced by Anti-miR33 in Atherosclerosis
- PMID: 26002865
- PMCID: PMC4578799
- DOI: 10.1161/CIRCRESAHA.117.305624
Macrophage Mitochondrial Energy Status Regulates Cholesterol Efflux and Is Enhanced by Anti-miR33 in Atherosclerosis
Abstract
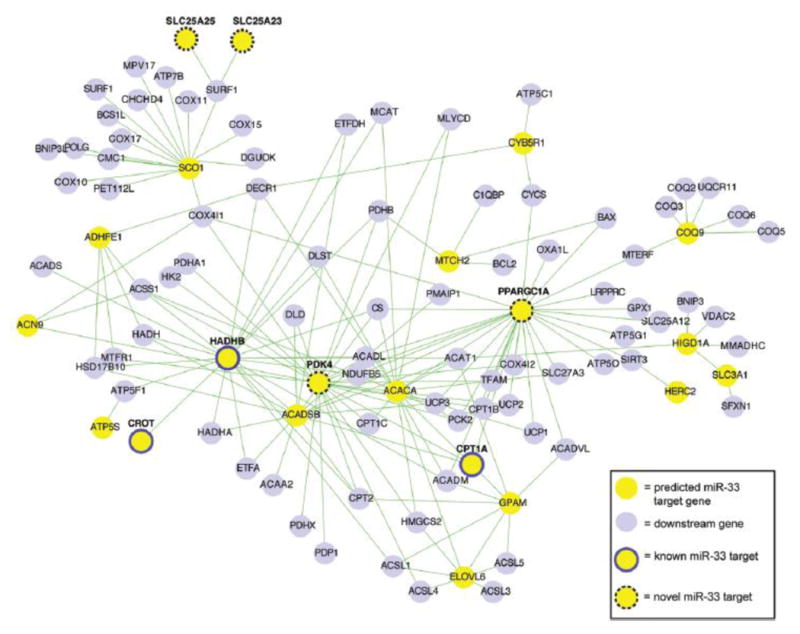
Rationale: Therapeutically targeting macrophage reverse cholesterol transport is a promising approach to treat atherosclerosis. Macrophage energy metabolism can significantly influence macrophage phenotype, but how this is controlled in foam cells is not known. Bioinformatic pathway analysis predicts that miR-33 represses a cluster of genes controlling cellular energy metabolism that may be important in macrophage cholesterol efflux.
Objective: We hypothesized that cellular energy status can influence cholesterol efflux from macrophages, and that miR-33 reduces cholesterol efflux via repression of mitochondrial energy metabolism pathways.
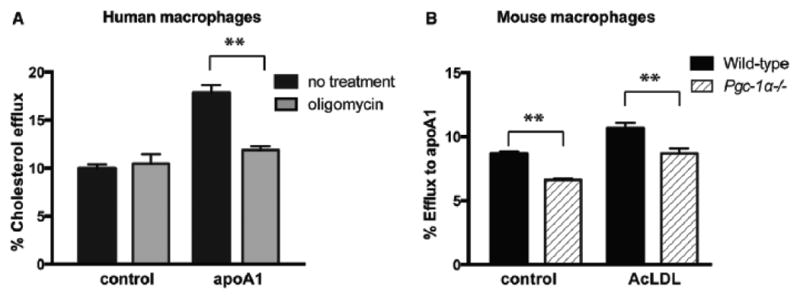
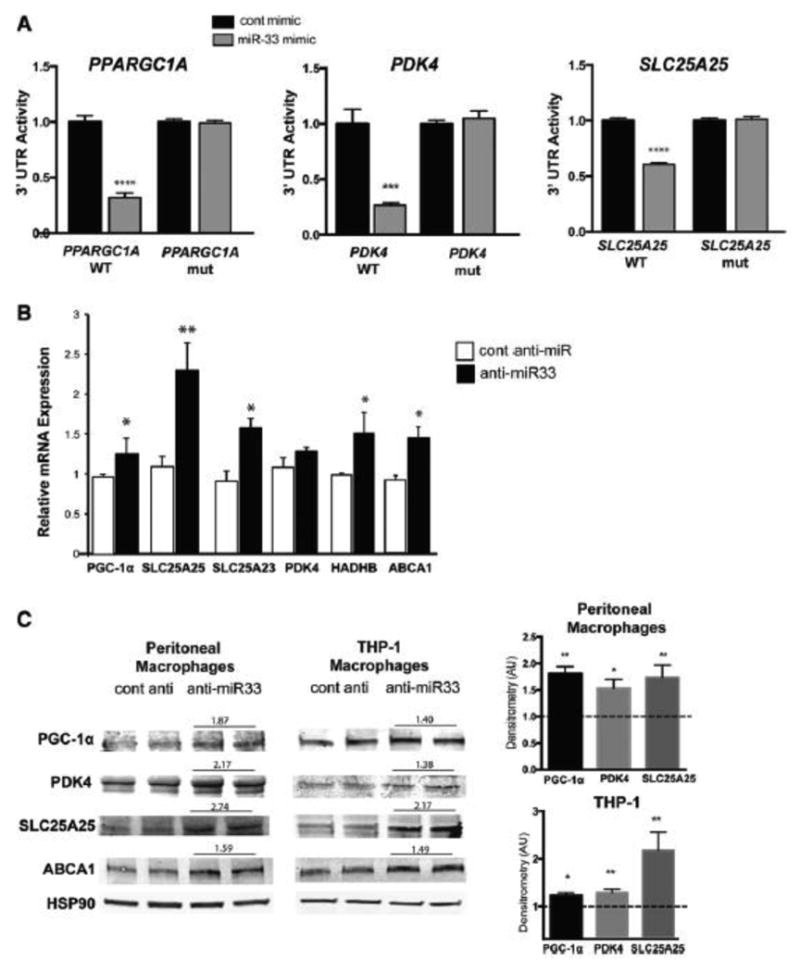
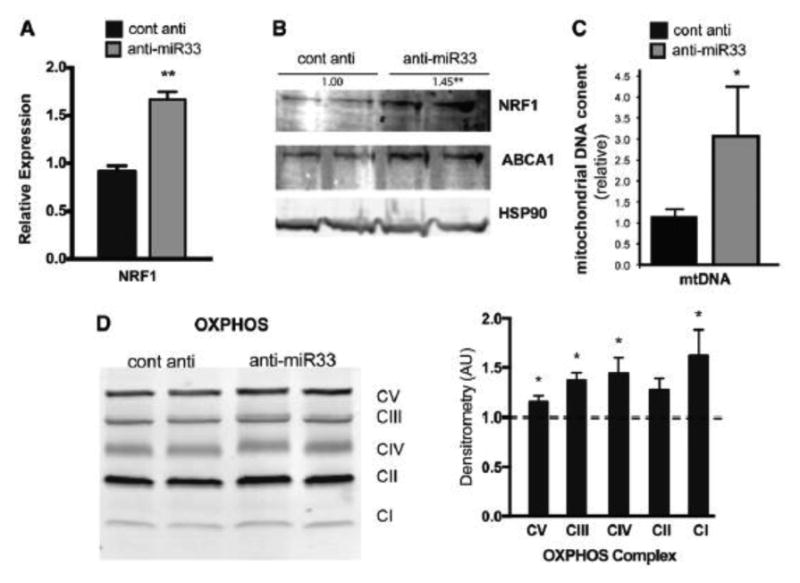
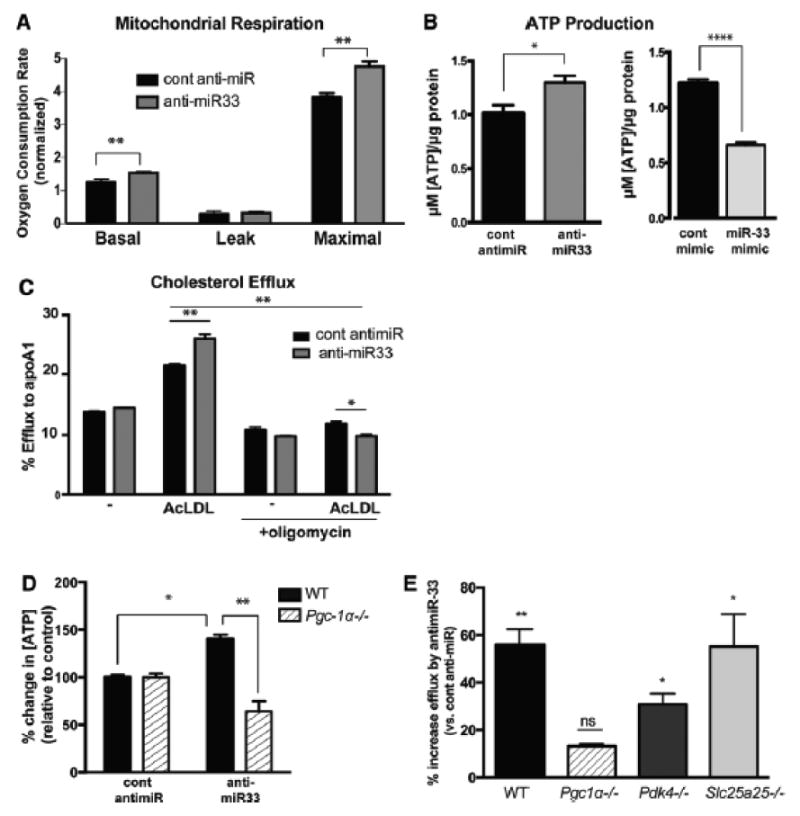
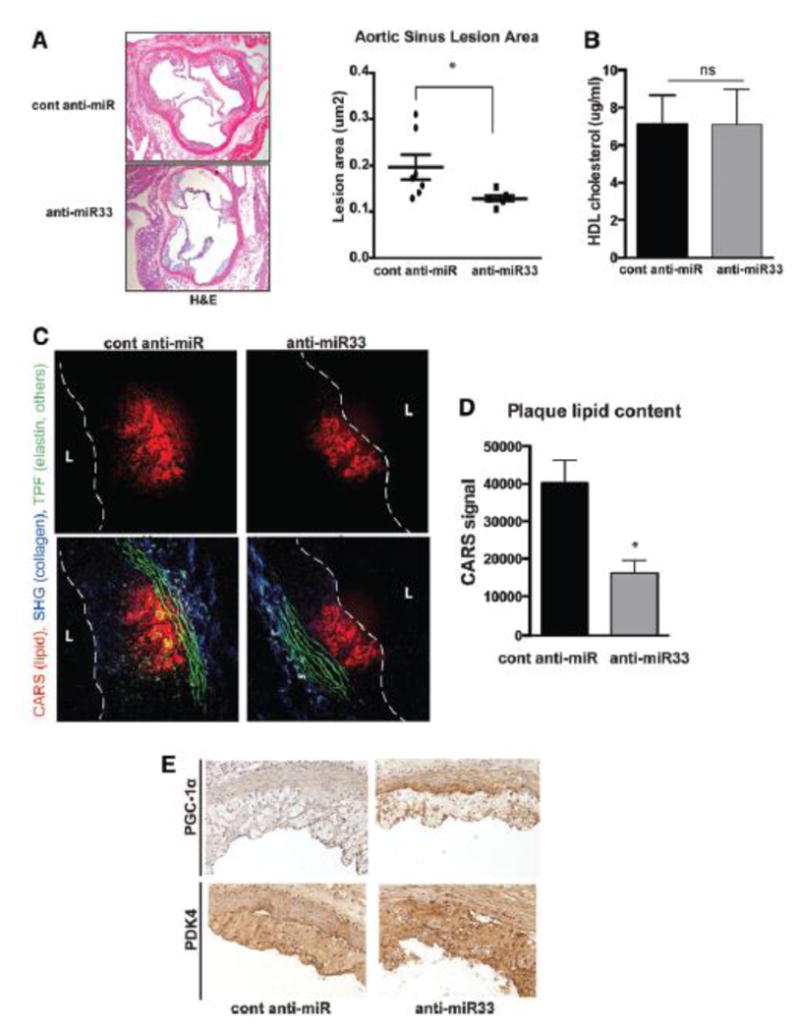
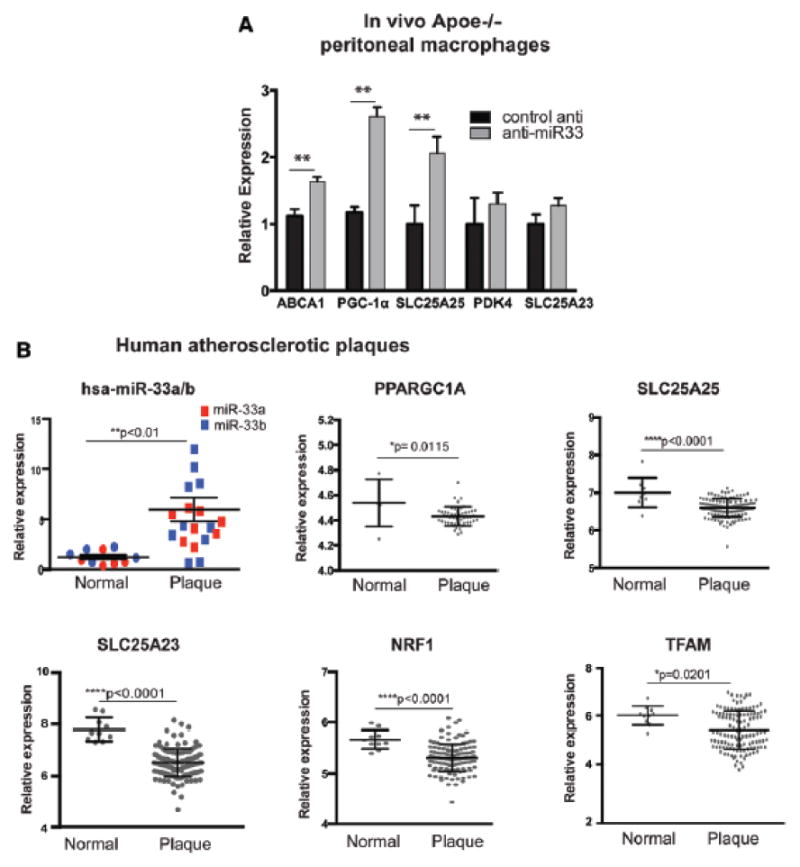
Methods and results: In this study, we demonstrated that macrophage cholesterol efflux is regulated by mitochondrial ATP production, and that miR-33 controls a network of genes that synchronize mitochondrial function. Inhibition of mitochondrial ATP synthase markedly reduces macrophage cholesterol efflux capacity, and anti-miR33 required fully functional mitochondria to enhance ABCA1-mediated cholesterol efflux. Specifically, anti-miR33 derepressed the novel target genes PGC-1α, PDK4, and SLC25A25 and boosted mitochondrial respiration and production of ATP. Treatment of atherosclerotic Apoe(-/-) mice with anti-miR33 oligonucleotides reduced aortic sinus lesion area compared with controls, despite no changes in high-density lipoprotein cholesterol or other circulating lipids. Expression of miR-33a/b was markedly increased in human carotid atherosclerotic plaques compared with normal arteries, and there was a concomitant decrease in mitochondrial regulatory genes PGC-1α, SLC25A25, NRF1, and TFAM, suggesting these genes are associated with advanced atherosclerosis in humans.
Conclusions: This study demonstrates that anti-miR33 therapy derepresses genes that enhance mitochondrial respiration and ATP production, which in conjunction with increased ABCA1 expression, works to promote macrophage cholesterol efflux and reduce atherosclerosis.
Keywords: atherosclerosis; cholesterol; macrophages; microRNA-33, mouse; mitochondria.
© 2015 American Heart Association, Inc.
Conflict of interest statement
Figures
Comment in
-
Novel Role of miR-33 in Regulating of Mitochondrial Function.Circ Res. 2015 Jul 17;117(3):225-8. doi: 10.1161/CIRCRESAHA.117.306949. Circ Res. 2015. PMID: 26185207 Free PMC article. No abstract available.
Similar articles
-
MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1.Atherosclerosis. 2014 Sep;236(1):215-26. doi: 10.1016/j.atherosclerosis.2014.07.005. Epub 2014 Jul 18. Atherosclerosis. 2014. PMID: 25084135
-
Diosgenin inhibits atherosclerosis via suppressing the MiR-19b-induced downregulation of ATP-binding cassette transporter A1.Atherosclerosis. 2015 May;240(1):80-9. doi: 10.1016/j.atherosclerosis.2015.02.044. Epub 2015 Feb 24. Atherosclerosis. 2015. PMID: 25765596
-
Novel Role of miR-33 in Regulating of Mitochondrial Function.Circ Res. 2015 Jul 17;117(3):225-8. doi: 10.1161/CIRCRESAHA.117.306949. Circ Res. 2015. PMID: 26185207 Free PMC article. No abstract available.
-
microRNAs in lipoprotein metabolism and cardiometabolic disorders.Atherosclerosis. 2016 Mar;246:352-60. doi: 10.1016/j.atherosclerosis.2016.01.025. Epub 2016 Jan 18. Atherosclerosis. 2016. PMID: 26828754 Free PMC article. Review.
-
From evolution to revolution: miRNAs as pharmacological targets for modulating cholesterol efflux and reverse cholesterol transport.Pharmacol Res. 2013 Sep;75:60-72. doi: 10.1016/j.phrs.2013.02.005. Epub 2013 Feb 19. Pharmacol Res. 2013. PMID: 23435093 Free PMC article. Review.
Cited by
-
Truths and controversies concerning the role of miRNAs in atherosclerosis and lipid metabolism.Curr Opin Lipidol. 2016 Dec;27(6):623-629. doi: 10.1097/MOL.0000000000000358. Curr Opin Lipidol. 2016. PMID: 27755115 Free PMC article. Review.
-
The Interplay between Oxidative Stress and miRNAs in Obesity-Associated Hepatic and Vascular Complications.Antioxidants (Basel). 2020 Jul 10;9(7):607. doi: 10.3390/antiox9070607. Antioxidants (Basel). 2020. PMID: 32664383 Free PMC article. Review.
-
The Role of Mitochondria-Targeting miRNAs in Intracerebral Hemorrhage.Curr Neuropharmacol. 2023;21(5):1065-1080. doi: 10.2174/1570159X20666220507021445. Curr Neuropharmacol. 2023. PMID: 35524670 Free PMC article. Review.
-
Insight into the Inter-Organ Crosstalk and Prognostic Role of Liver-Derived MicroRNAs in Metabolic Disease Progression.Biomedicines. 2023 May 31;11(6):1597. doi: 10.3390/biomedicines11061597. Biomedicines. 2023. PMID: 37371692 Free PMC article. Review.
-
Myeloid BAF60a deficiency alters metabolic homeostasis and exacerbates atherosclerosis.Cell Rep. 2023 Oct 31;42(10):113171. doi: 10.1016/j.celrep.2023.113171. Epub 2023 Sep 27. Cell Rep. 2023. PMID: 37768825 Free PMC article.
References
-
- Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–6. - PubMed
-
- Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19. - PMC - PubMed
-
- Allen AM, Taylor JM, Graham A. Mitochondrial (dys)function and regulation of macrophage cholesterol efflux. Clin Sci (Lond) 2013;124:509–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
