

Coupling the CRISPR/Cas9 System with Lambda Red Recombineering Enables Simplified Chromosomal Gene Replacement in Escherichia coli
- PMID: 26002895
- PMCID: PMC4495200
- DOI: 10.1128/AEM.01248-15
Coupling the CRISPR/Cas9 System with Lambda Red Recombineering Enables Simplified Chromosomal Gene Replacement in Escherichia coli
Abstract
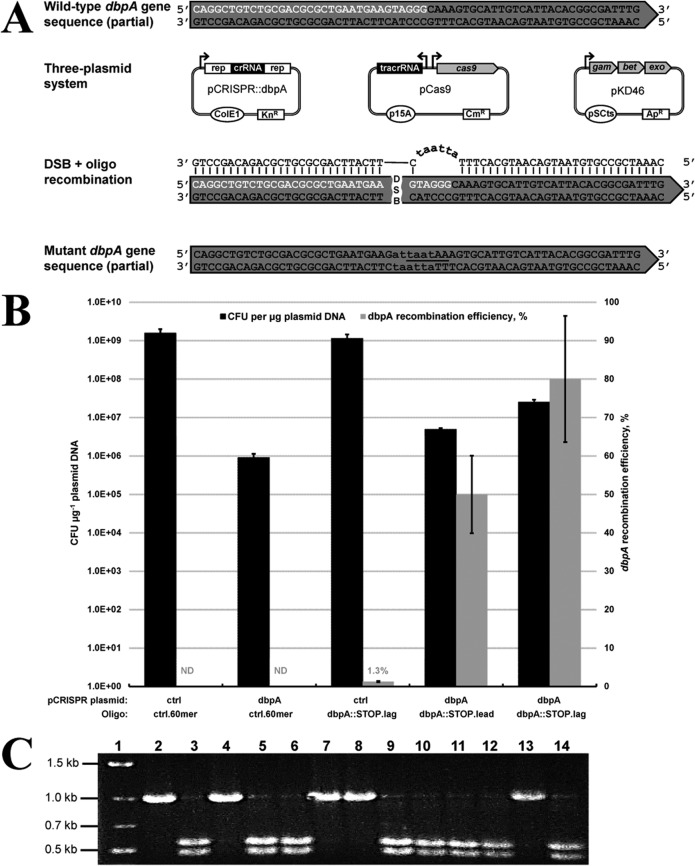
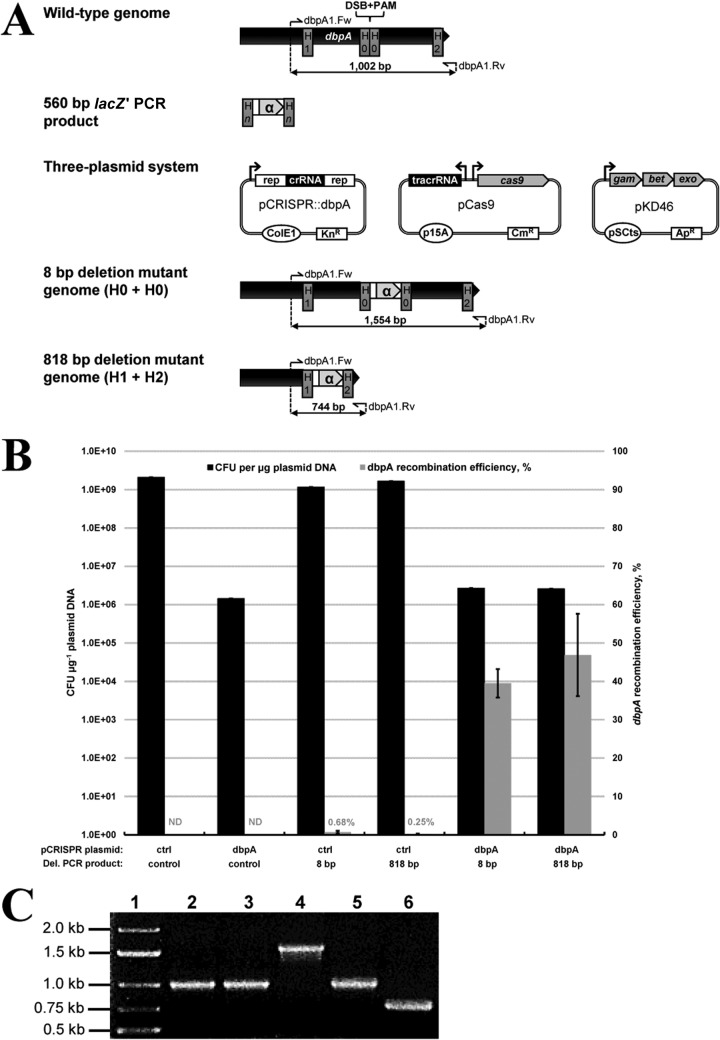
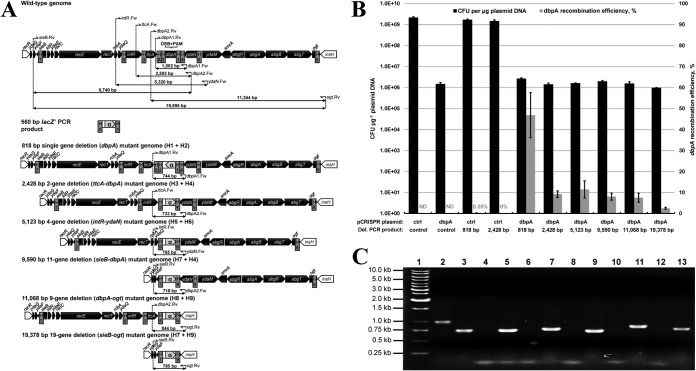
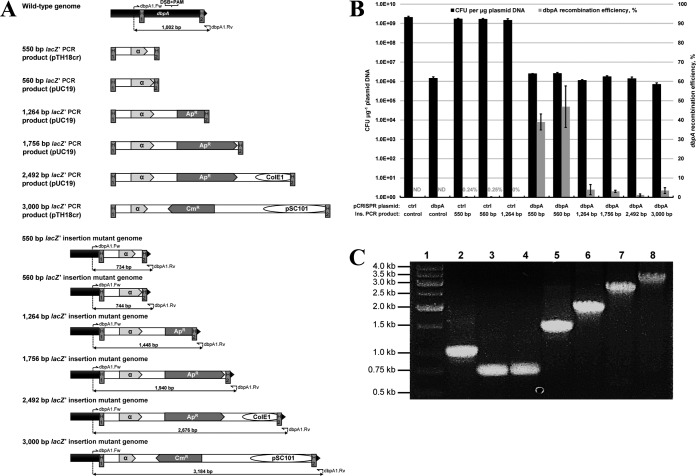
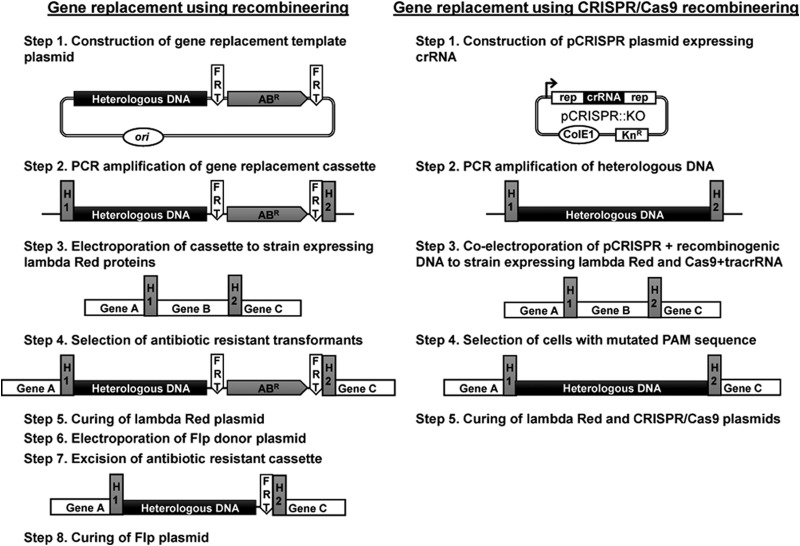
To date, most genetic engineering approaches coupling the type II Streptococcus pyogenes clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system to lambda Red recombineering have involved minor single nucleotide mutations. Here we show that procedures for carrying out more complex chromosomal gene replacements in Escherichia coli can be substantially enhanced through implementation of CRISPR/Cas9 genome editing. We developed a three-plasmid approach that allows not only highly efficient recombination of short single-stranded oligonucleotides but also replacement of multigene chromosomal stretches of DNA with large PCR products. By systematically challenging the proposed system with respect to the magnitude of chromosomal deletion and size of DNA insertion, we demonstrated DNA deletions of up to 19.4 kb, encompassing 19 nonessential chromosomal genes, and insertion of up to 3 kb of heterologous DNA with recombination efficiencies permitting mutant detection by colony PCR screening. Since CRISPR/Cas9-coupled recombineering does not rely on the use of chromosome-encoded antibiotic resistance, or flippase recombination for antibiotic marker recycling, our approach is simpler, less labor-intensive, and allows efficient production of gene replacement mutants that are both markerless and "scar"-less.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Expression of Shewanella frigidimarina fatty acid metabolic genes in E. coli by CRISPR/cas9-coupled lambda Red recombineering.Biotechnol Lett. 2016 Jan;38(1):117-22. doi: 10.1007/s10529-015-1956-4. Epub 2015 Sep 10. Biotechnol Lett. 2016. PMID: 26358622
-
The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli.Sci Rep. 2015 Oct 14;5:15096. doi: 10.1038/srep15096. Sci Rep. 2015. PMID: 26463009 Free PMC article.
-
One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces.Acta Biochim Biophys Sin (Shanghai). 2015 Apr;47(4):231-43. doi: 10.1093/abbs/gmv007. Epub 2015 Mar 3. Acta Biochim Biophys Sin (Shanghai). 2015. PMID: 25739462
-
Recombineering: genetic engineering in bacteria using homologous recombination.Curr Protoc Mol Biol. 2007 Apr;Chapter 1:Unit 1.16. doi: 10.1002/0471142727.mb0116s78. Curr Protoc Mol Biol. 2007. PMID: 18265390 Review.
-
[Recombineering and its application].Yi Chuan Xue Bao. 2003 Oct;30(10):983-8. Yi Chuan Xue Bao. 2003. PMID: 14669518 Review. Chinese.
Cited by
-
Development of a CRISPR-Cas9 Tool Kit for Comprehensive Engineering of Bacillus subtilis.Appl Environ Microbiol. 2016 Jul 29;82(16):4876-95. doi: 10.1128/AEM.01159-16. Print 2016 Aug 15. Appl Environ Microbiol. 2016. PMID: 27260361 Free PMC article.
-
Engineering of the Small Noncoding RNA (sRNA) DsrA Together with the sRNA Chaperone Hfq Enhances the Acid Tolerance of Escherichia coli.Appl Environ Microbiol. 2021 Apr 27;87(10):e02923-20. doi: 10.1128/AEM.02923-20. Print 2021 Apr 27. Appl Environ Microbiol. 2021. PMID: 33674434 Free PMC article.
-
Precision design of stable genetic circuits carried in highly-insulated E. coli genomic landing pads.Mol Syst Biol. 2020 Aug;16(8):e9584. doi: 10.15252/msb.20209584. Mol Syst Biol. 2020. PMID: 32812710 Free PMC article.
-
Multiple-step chromosomal integration of divided segments from a large DNA fragment via CRISPR/Cas9 in Escherichia coli.J Ind Microbiol Biotechnol. 2019 Jan;46(1):81-90. doi: 10.1007/s10295-018-2114-5. Epub 2018 Nov 23. J Ind Microbiol Biotechnol. 2019. PMID: 30470963
-
Coupling CRISPR/Cas9 and Lambda Red Recombineering System for Genome Editing of Salmonella Gallinarum and the Effect of ssaU Knock-Out Mutant on the Virulence of Bacteria.Biomedicines. 2022 Nov 24;10(12):3028. doi: 10.3390/biomedicines10123028. Biomedicines. 2022. PMID: 36551784 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
