

DNA Demethylation Dynamics in the Human Prenatal Germline
- PMID: 26004067
- PMCID: PMC4458157
- DOI: 10.1016/j.cell.2015.05.012
DNA Demethylation Dynamics in the Human Prenatal Germline
Abstract
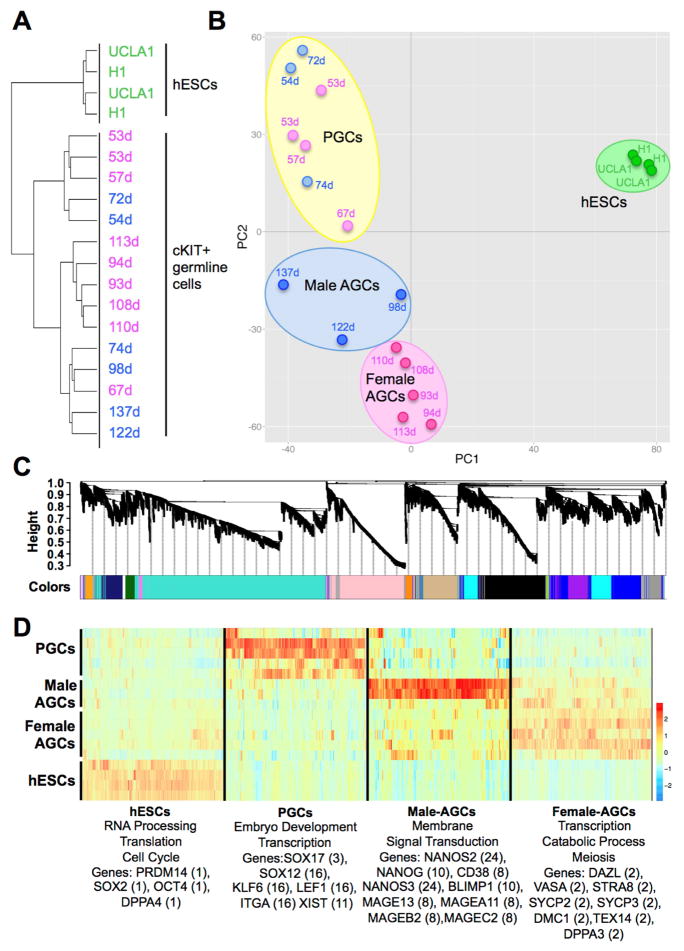
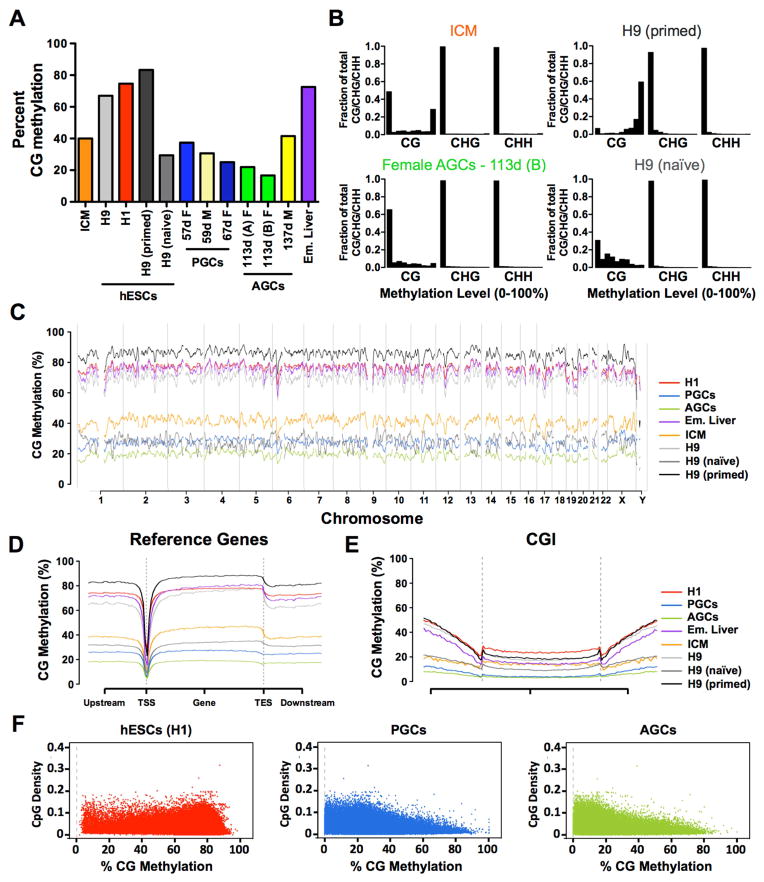
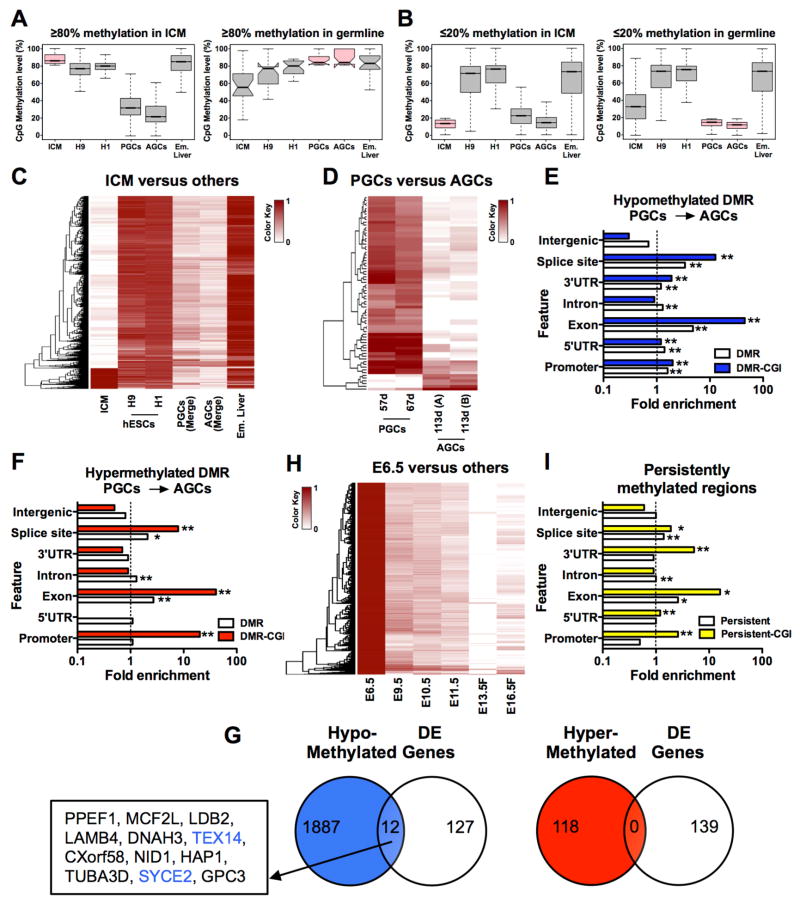
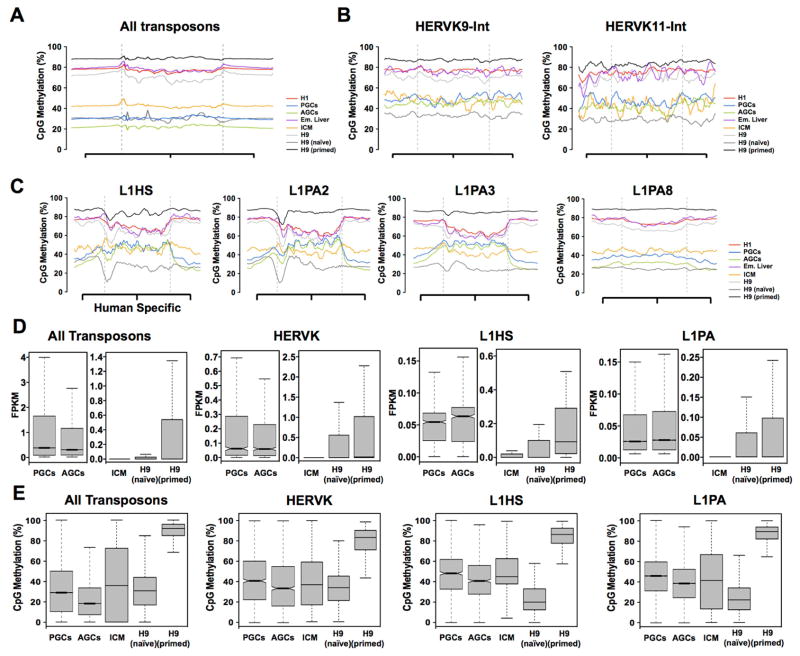
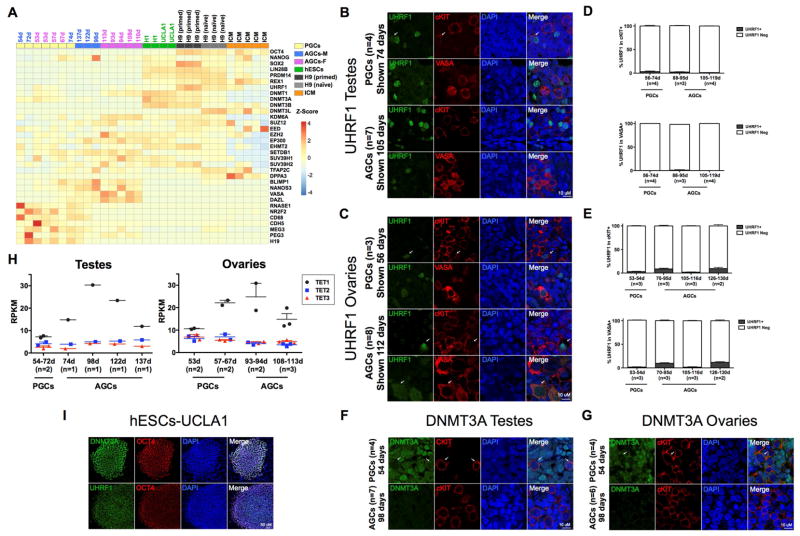
Global DNA demethylation in humans is a fundamental process that occurs in pre-implantation embryos and reversion to naive ground state pluripotent stem cells (PSCs). However, the extent of DNA methylation reprogramming in human germline cells is unknown. Here, we performed whole-genome bisulfite sequencing (WGBS) and RNA-sequencing (RNA-seq) of human prenatal germline cells from 53 to 137 days of development. We discovered that the transcriptome and methylome of human germline is distinct from both human PSCs and the inner cell mass (ICM) of human blastocysts. Using this resource to monitor the outcome of global DNA demethylation with reversion of primed PSCs to the naive ground state, we uncovered hotspots of ultralow methylation at transposons that are protected from demethylation in the germline and ICM. Taken together, the human germline serves as a valuable in vivo tool for monitoring the epigenome of cells that have emerged from a global DNA demethylation event.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
References
-
- Aramaki S, et al. A mesodermal factor, T, specifies mouse germ cell fate by directly activating germline determinants. Dev Cell. 2013;27:516–529. - PubMed
-
- Chan YS, Göke J, Ng JH, Lu X, Gonzales KA, Tan CP, Tng WQ, Hong ZZ, Lim YS, Ng HH. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell. 2013;13:663–675. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
