

The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration
- PMID: 26005849
- PMCID: PMC4482801
- DOI: 10.1038/nn.4024
The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration
Abstract
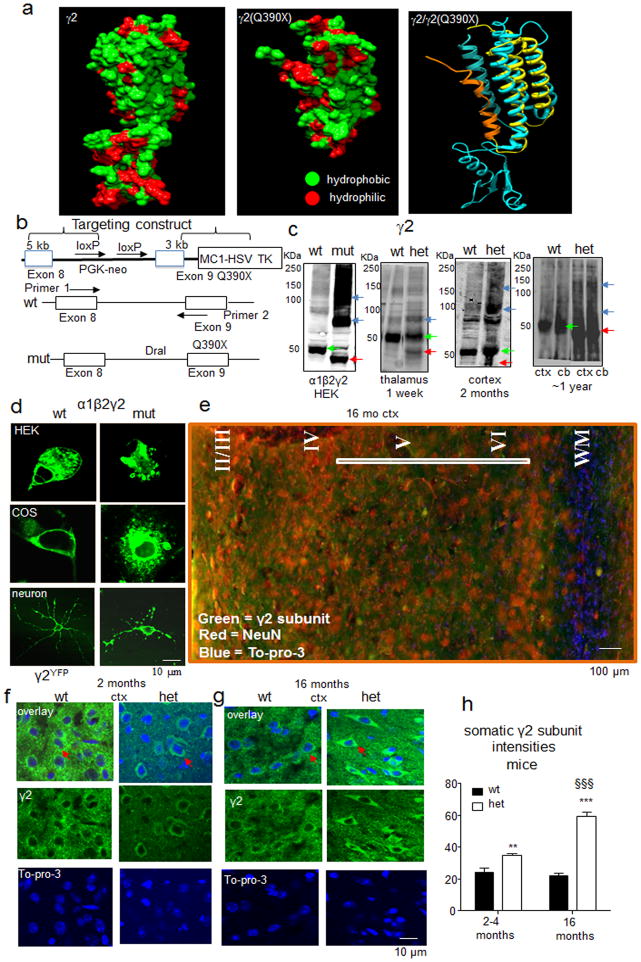
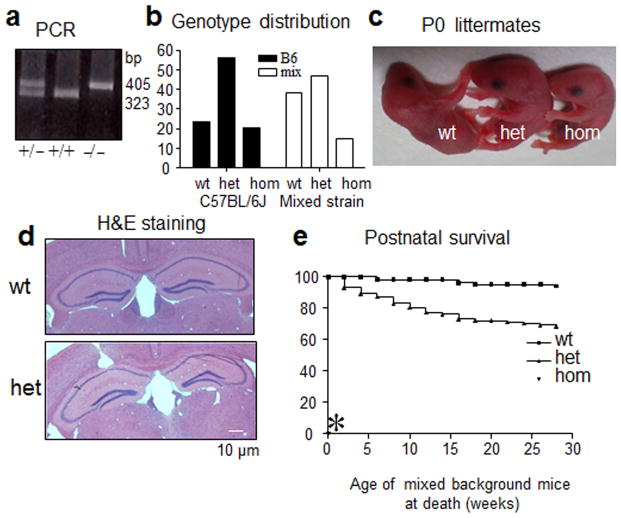
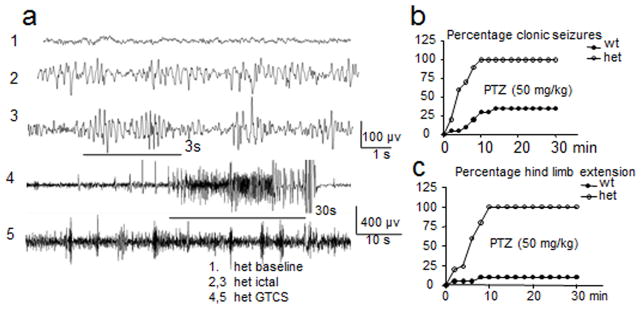
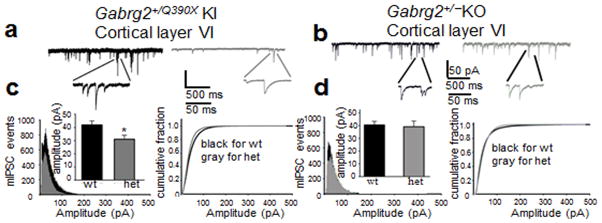
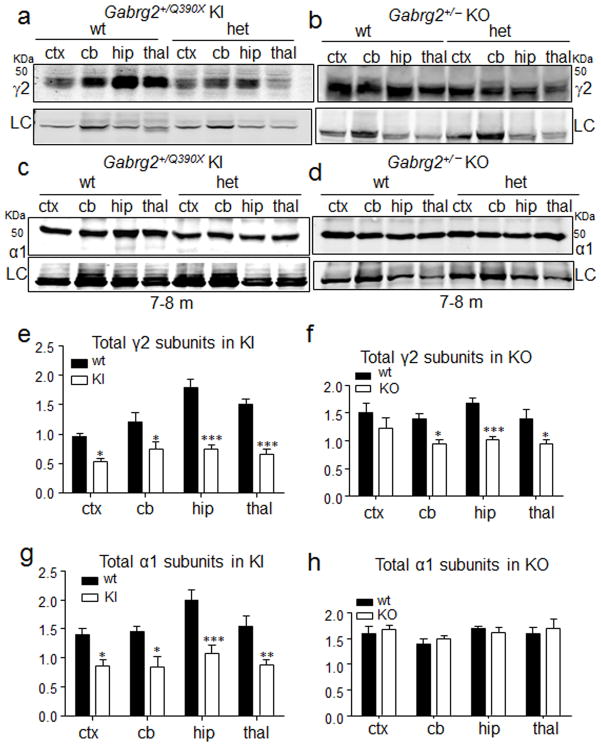
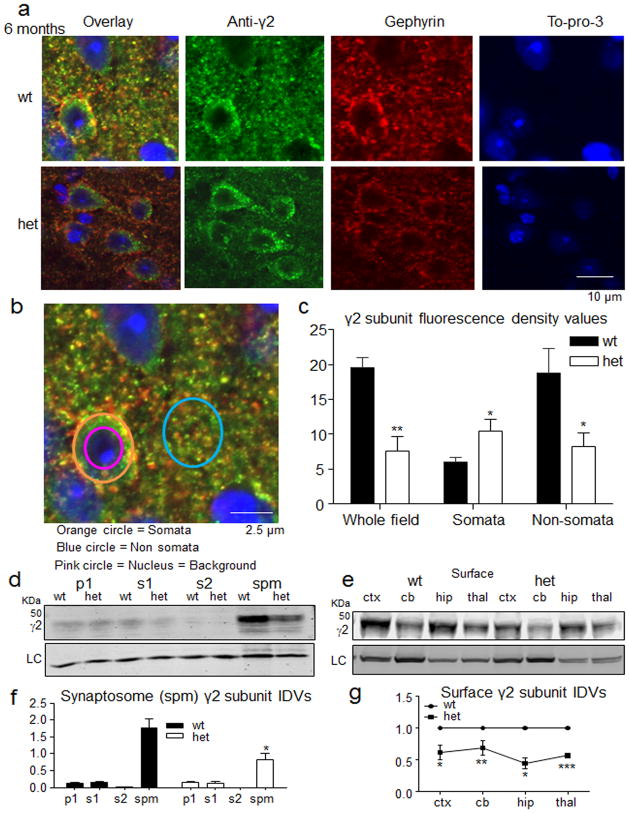
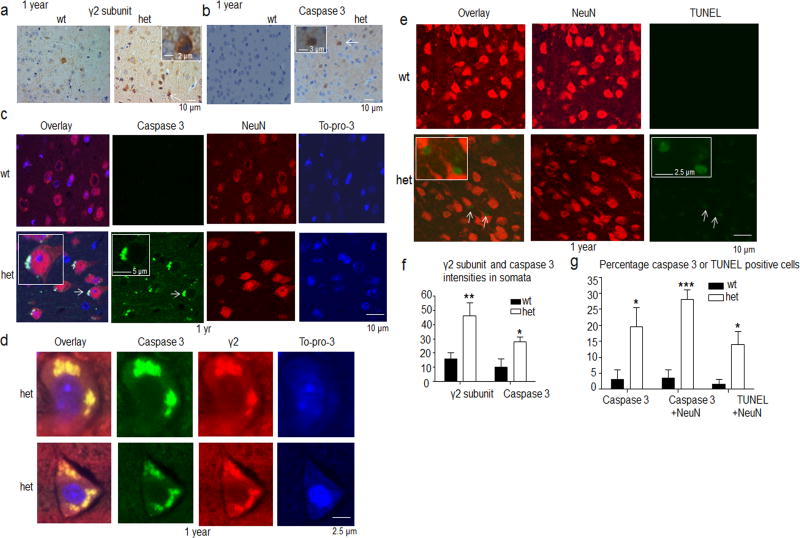
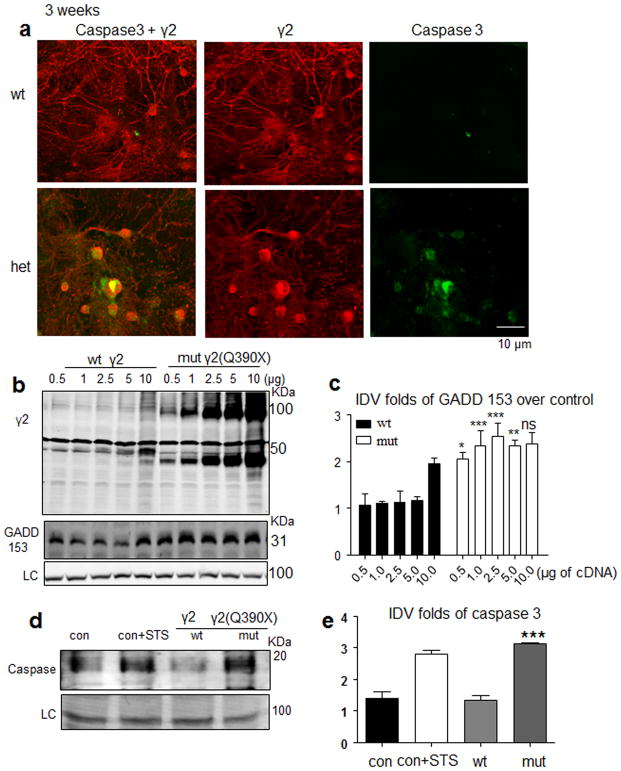
Genetic epilepsy and neurodegenerative diseases are two common neurological disorders that are conventionally viewed as being unrelated. A subset of patients with severe genetic epilepsies who have impaired development and often go on to die of their disease respond poorly to anticonvulsant drug therapy, suggesting a need for new therapeutic targets. Previously, we reported that multiple GABAA receptor epilepsy mutations result in protein misfolding and abnormal receptor trafficking. We have now developed a model of a severe human genetic epileptic encephalopathy, the Gabrg2(+/Q390X) knock-in mouse. We found that, in addition to impairing inhibitory neurotransmission, mutant GABAA receptor γ2(Q390X) subunits accumulated and aggregated intracellularly, activated caspase 3 and caused widespread, age-dependent neurodegeneration. These findings suggest that the fundamental protein metabolism and cellular consequences of the epilepsy-associated mutant γ2(Q390X) ion channel subunit are not fundamentally different from those associated with neurodegeneration. Our results have far-reaching relevance for the identification of conserved pathological cascades and mechanism-based therapies that are shared between genetic epilepsies and neurodegenerative diseases.
Figures
Similar articles
-
Overexpressing wild-type γ2 subunits rescued the seizure phenotype in Gabrg2+/Q390X Dravet syndrome mice.Epilepsia. 2017 Aug;58(8):1451-1461. doi: 10.1111/epi.13810. Epub 2017 Jun 6. Epilepsia. 2017. PMID: 28586508 Free PMC article.
-
4-Phenylbutyrate promoted wild-type γ-aminobutyric acid type A receptor trafficking, reduced endoplasmic reticulum stress, and mitigated seizures in Gabrg2+/Q390X mice associated with Dravet syndrome.Epilepsia. 2024 Jan;65(1):204-217. doi: 10.1111/epi.17779. Epub 2023 Nov 29. Epilepsia. 2024. PMID: 37746768 Free PMC article.
-
Altered GABAA receptor expression in brainstem nuclei and SUDEP in Gabrg2(+/Q390X) mice associated with epileptic encephalopathy.Epilepsy Res. 2016 Jul;123:50-4. doi: 10.1016/j.eplepsyres.2016.04.002. Epub 2016 Apr 13. Epilepsy Res. 2016. PMID: 27131289 Free PMC article.
-
Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome.JAMA Neurol. 2016 Aug 1;73(8):1009-16. doi: 10.1001/jamaneurol.2016.0449. JAMA Neurol. 2016. PMID: 27367160 Free PMC article. Review.
-
Mutations in GABAA receptor subunits associated with genetic epilepsies.J Physiol. 2010 Jun 1;588(Pt 11):1861-9. doi: 10.1113/jphysiol.2010.186999. Epub 2010 Mar 22. J Physiol. 2010. PMID: 20308251 Free PMC article. Review.
Cited by
-
Genetic mosaicism, intrafamilial phenotypic heterogeneity, and molecular defects of a novel missense SLC6A1 mutation associated with epilepsy and ADHD.Exp Neurol. 2021 Aug;342:113723. doi: 10.1016/j.expneurol.2021.113723. Epub 2021 May 5. Exp Neurol. 2021. PMID: 33961861 Free PMC article.
-
Differential protein structural disturbances and suppression of assembly partners produced by nonsense GABRG2 epilepsy mutations: implications for disease phenotypic heterogeneity.Sci Rep. 2016 Oct 20;6:35294. doi: 10.1038/srep35294. Sci Rep. 2016. PMID: 27762395 Free PMC article.
-
Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype.Int J Mol Sci. 2022 Aug 26;23(17):9683. doi: 10.3390/ijms23179683. Int J Mol Sci. 2022. PMID: 36077081 Free PMC article. Review.
-
Disruption of the Ubiquitin-Proteasome System and Elevated Endoplasmic Reticulum Stress in Epilepsy.Biomedicines. 2022 Mar 11;10(3):647. doi: 10.3390/biomedicines10030647. Biomedicines. 2022. PMID: 35327449 Free PMC article. Review.
-
Overexpressing wild-type γ2 subunits rescued the seizure phenotype in Gabrg2+/Q390X Dravet syndrome mice.Epilepsia. 2017 Aug;58(8):1451-1461. doi: 10.1111/epi.13810. Epub 2017 Jun 6. Epilepsia. 2017. PMID: 28586508 Free PMC article.
References
-
- Noebels JL. Exploring new gene discoveries in idiopathic generalized epilepsy. Epilepsia. 2003;44(Suppl 2):16–21. - PubMed
-
- Heron SE, et al. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin. J Med Genet. 2010;47:137–141. - PubMed
-
- Shi X, et al. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 2009;31:758–762. - PubMed
-
- Ogiwara I, et al. A homozygous mutation of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia. 2012;53:e200–e203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
