

Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β
- PMID: 26008898
- PMCID: PMC4451132
- DOI: 10.1084/jem.20142384
Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β
Abstract
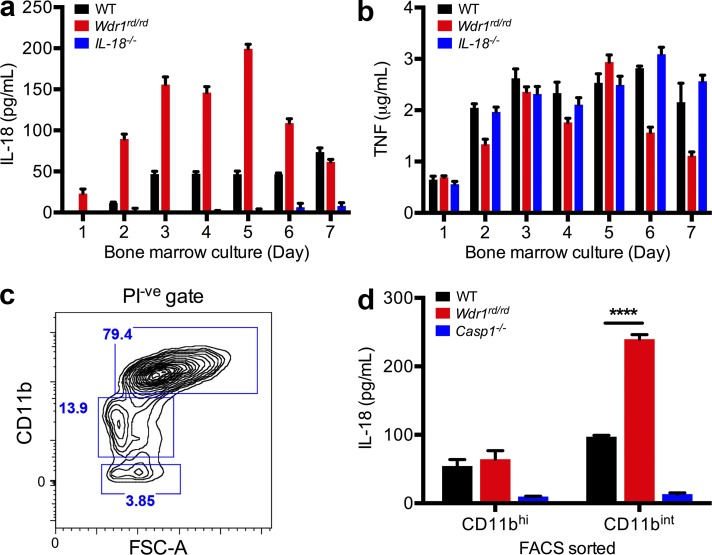
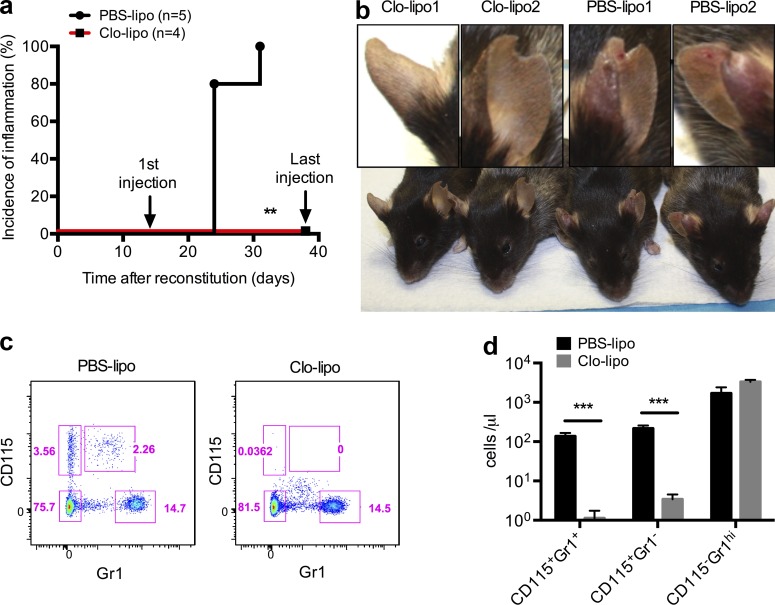
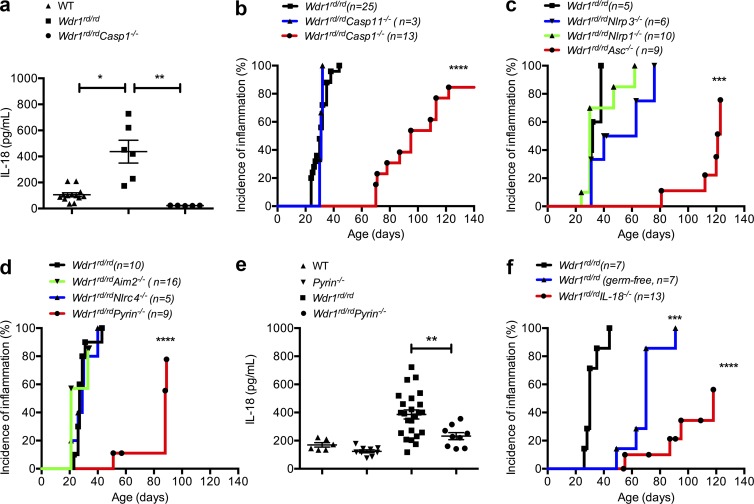
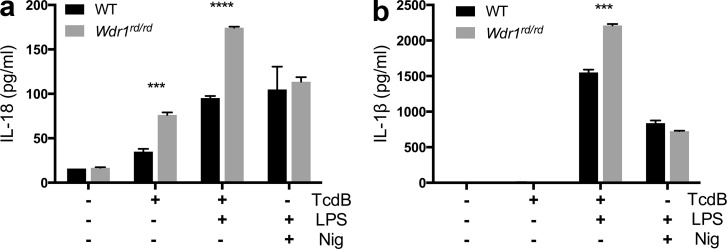
Gain-of-function mutations that activate the innate immune system can cause systemic autoinflammatory diseases associated with increased IL-1β production. This cytokine is activated identically to IL-18 by an intracellular protein complex known as the inflammasome; however, IL-18 has not yet been specifically implicated in the pathogenesis of hereditary autoinflammatory disorders. We have now identified an autoinflammatory disease in mice driven by IL-18, but not IL-1β, resulting from an inactivating mutation of the actin-depolymerizing cofactor Wdr1. This perturbation of actin polymerization leads to systemic autoinflammation that is reduced when IL-18 is deleted but not when IL-1 signaling is removed. Remarkably, inflammasome activation in mature macrophages is unaltered, but IL-18 production from monocytes is greatly exaggerated, and depletion of monocytes in vivo prevents the disease. Small-molecule inhibition of actin polymerization can remove potential danger signals from the system and prevents monocyte IL-18 production. Finally, we show that the inflammasome sensor of actin dynamics in this system requires caspase-1, apoptosis-associated speck-like protein containing a caspase recruitment domain, and the innate immune receptor pyrin. Previously, perturbation of actin polymerization by pathogens was shown to activate the pyrin inflammasome, so our data now extend this guard hypothesis to host-regulated actin-dependent processes and autoinflammatory disease.
© 2015 Kim et al.
Figures




Comment in
-
Inflammation on the move.J Exp Med. 2015 Jun 1;212(6):829-30. doi: 10.1084/jem.2126insight3. J Exp Med. 2015. PMID: 26034116 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
