

Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia
- PMID: 26008899
- PMCID: PMC4451137
- DOI: 10.1084/jem.20141130
Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia
Abstract
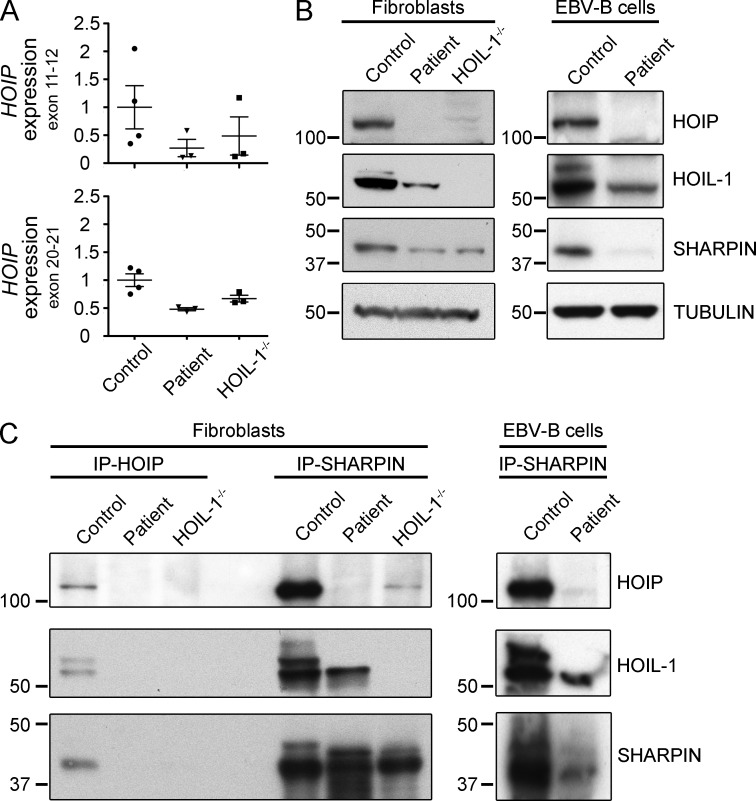
Inherited, complete deficiency of human HOIL-1, a component of the linear ubiquitination chain assembly complex (LUBAC), underlies autoinflammation, infections, and amylopectinosis. We report the clinical description and molecular analysis of a novel inherited disorder of the human LUBAC complex. A patient with multiorgan autoinflammation, combined immunodeficiency, subclinical amylopectinosis, and systemic lymphangiectasia, is homozygous for a mutation in HOIP, the gene encoding the catalytic component of LUBAC. The missense allele (L72P, in the PUB domain) is at least severely hypomorphic, as it impairs HOIP expression and destabilizes the whole LUBAC complex. Linear ubiquitination and NF-κB activation are impaired in the patient's fibroblasts stimulated by IL-1β or TNF. In contrast, the patient's monocytes respond to IL-1β more vigorously than control monocytes. However, the activation and differentiation of the patient's B cells are impaired in response to CD40 engagement. These cellular and clinical phenotypes largely overlap those of HOIL-1-deficient patients. Clinical differences between HOIL-1- and HOIP-mutated patients may result from differences between the mutations, the loci, or other factors. Our findings show that human HOIP is essential for the assembly and function of LUBAC and for various processes governing inflammation and immunity in both hematopoietic and nonhematopoietic cells.
© 2015 Boisson et al.
Figures





References
-
- Al-Herz W., Bousfiha A., Casanova J.L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Franco J.L., Gaspar H.B., Holland S.M., et al. 2014. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front. Immunol. 5:162 10.3389/fimmu.2014.00162 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
