

Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment
- PMID: 26009596
- PMCID: PMC4576143
- DOI: 10.1093/eurheartj/ehv157
Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment
Abstract
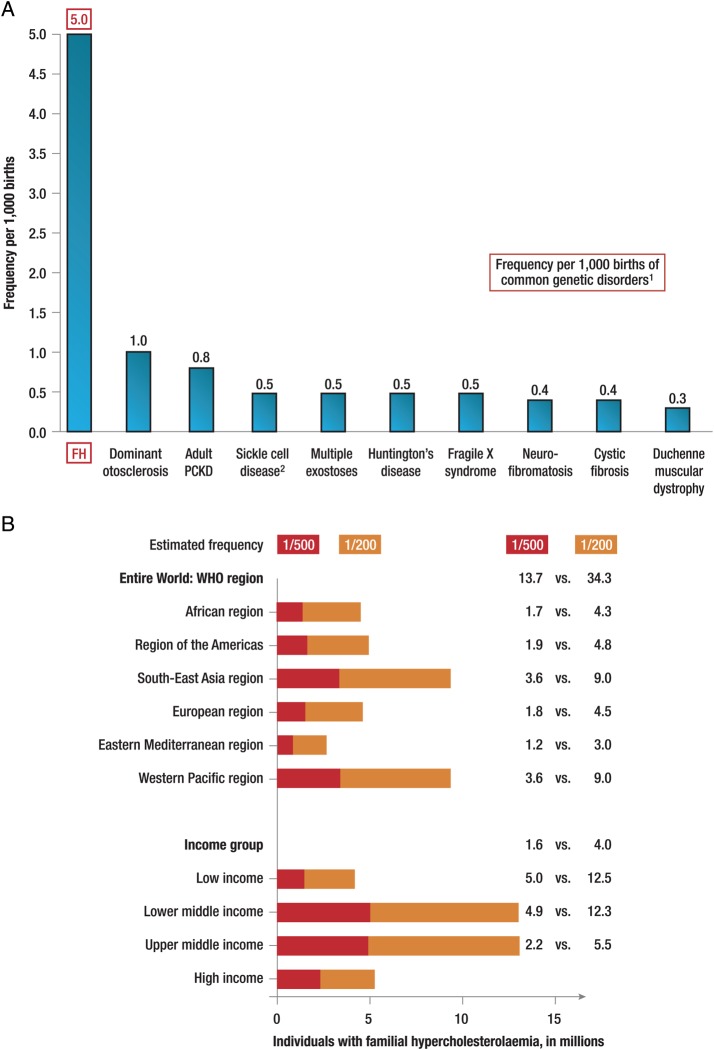
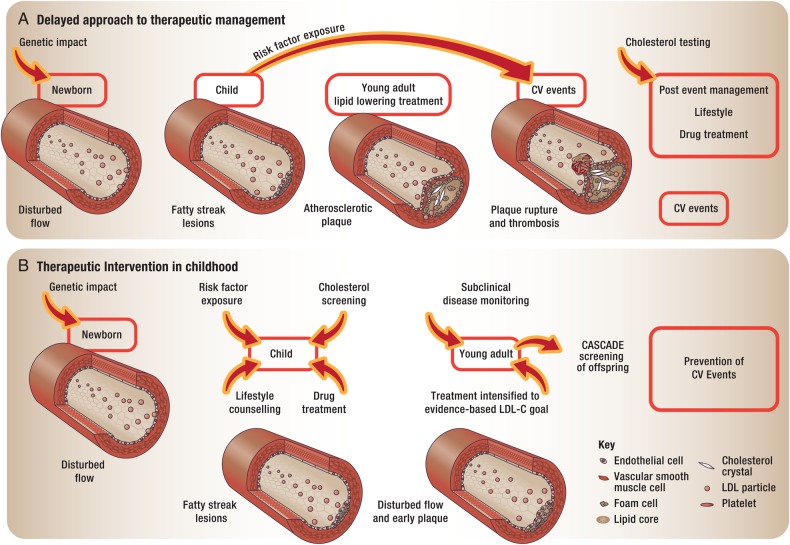
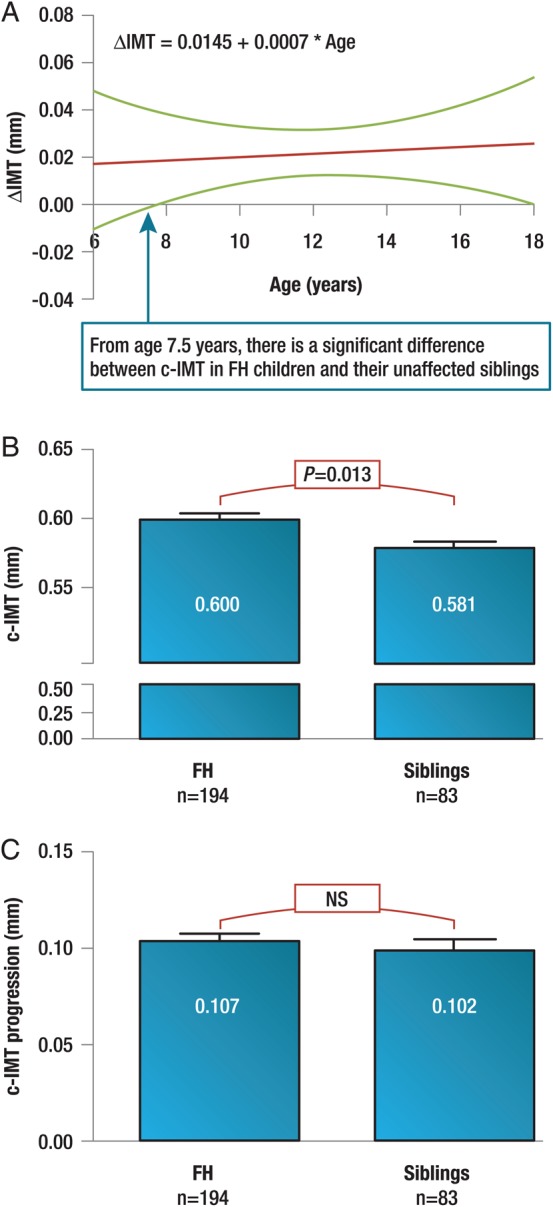
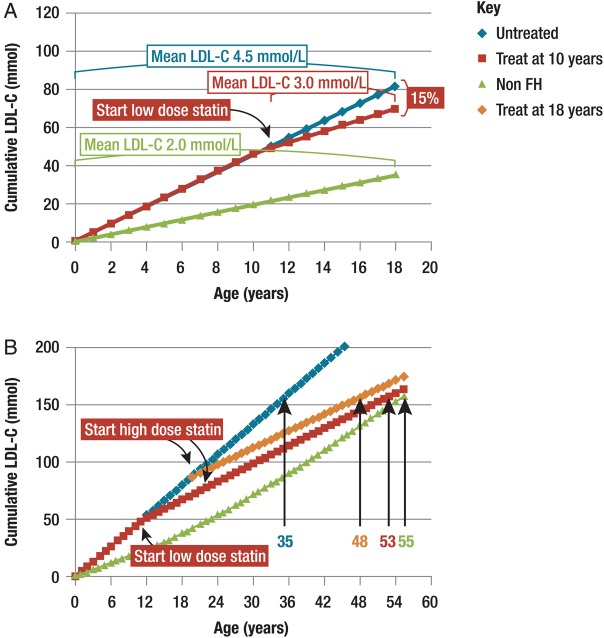
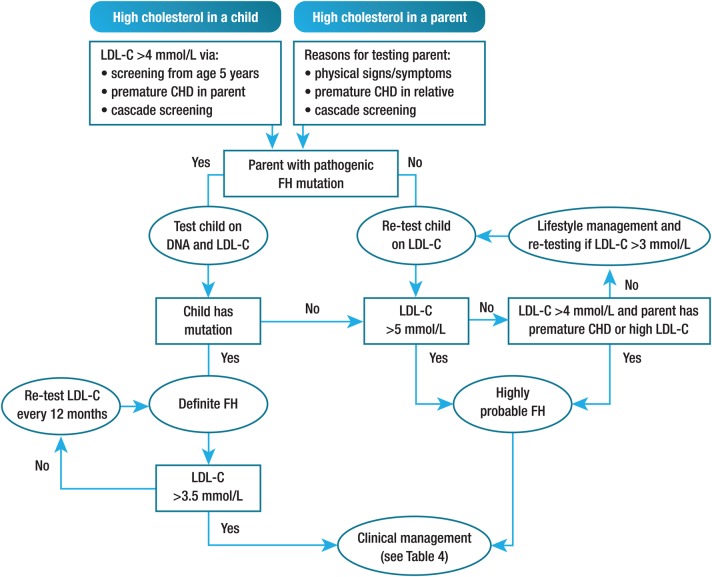
Familial hypercholesterolaemia (FH) is a common genetic cause of premature coronary heart disease (CHD). Globally, one baby is born with FH every minute. If diagnosed and treated early in childhood, individuals with FH can have normal life expectancy. This consensus paper aims to improve awareness of the need for early detection and management of FH children. Familial hypercholesterolaemia is diagnosed either on phenotypic criteria, i.e. an elevated low-density lipoprotein cholesterol (LDL-C) level plus a family history of elevated LDL-C, premature coronary artery disease and/or genetic diagnosis, or positive genetic testing. Childhood is the optimal period for discrimination between FH and non-FH using LDL-C screening. An LDL-C ≥5 mmol/L (190 mg/dL), or an LDL-C ≥4 mmol/L (160 mg/dL) with family history of premature CHD and/or high baseline cholesterol in one parent, make the phenotypic diagnosis. If a parent has a genetic defect, the LDL-C cut-off for the child is ≥3.5 mmol/L (130 mg/dL). We recommend cascade screening of families using a combined phenotypic and genotypic strategy. In children, testing is recommended from age 5 years, or earlier if homozygous FH is suspected. A healthy lifestyle and statin treatment (from age 8 to 10 years) are the cornerstones of management of heterozygous FH. Target LDL-C is <3.5 mmol/L (130 mg/dL) if >10 years, or ideally 50% reduction from baseline if 8-10 years, especially with very high LDL-C, elevated lipoprotein(a), a family history of premature CHD or other cardiovascular risk factors, balanced against the long-term risk of treatment side effects. Identifying FH early and optimally lowering LDL-C over the lifespan reduces cumulative LDL-C burden and offers health and socioeconomic benefits. To drive policy change for timely detection and management, we call for further studies in the young. Increased awareness, early identification, and optimal treatment from childhood are critical to adding decades of healthy life for children and adolescents with FH.
Keywords: Adolescents; Children; Consensus statement; Diagnosis; Ezetimibe; Familial hypercholesterolaemia; LDL cholesterol; PCSK9 inhibitor; Statin; Treatment.
© The Author 2015. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures
References
-
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986;232:34–47. - PubMed
-
- Sjouke B, Kusters DM, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, Roeters van Lennep JE, Stalenhoef AF, Wiegman A, de Graaf J, Fouchier SW, Kastelein JJ, Hovingh G. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J 2015;36:560–565. - PubMed
-
- Brown MS, Kovanen PT, Goldstein JL, Eeckels R, Vandenberghe K, van den Berghe H, Fryns JP, Cassiman JJ. Prenatal diagnosis of homozygous familial hypercholesterolaemia. Expression of a genetic receptor disease in utero. Lancet 1978;1:526–529. - PubMed
-
- Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Kees Hovingh G, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–3490a. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
