

Identification of B6SJL mSOD1(G93A) mouse subgroups with different disease progression rates
- PMID: 26010802
- PMCID: PMC4607568
- DOI: 10.1002/cne.23814
Identification of B6SJL mSOD1(G93A) mouse subgroups with different disease progression rates
Abstract
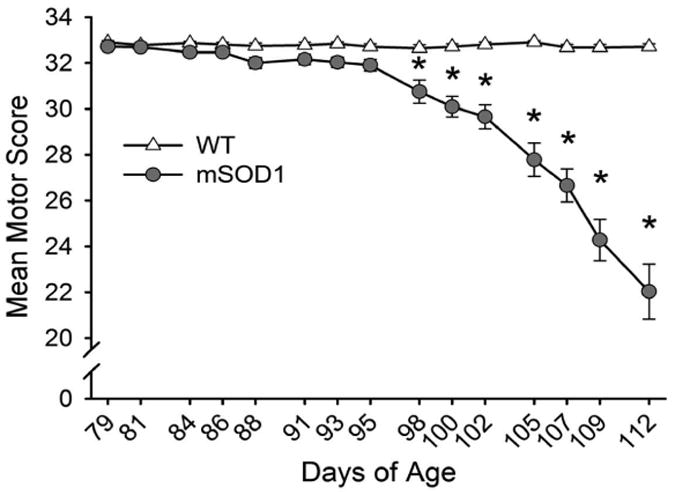
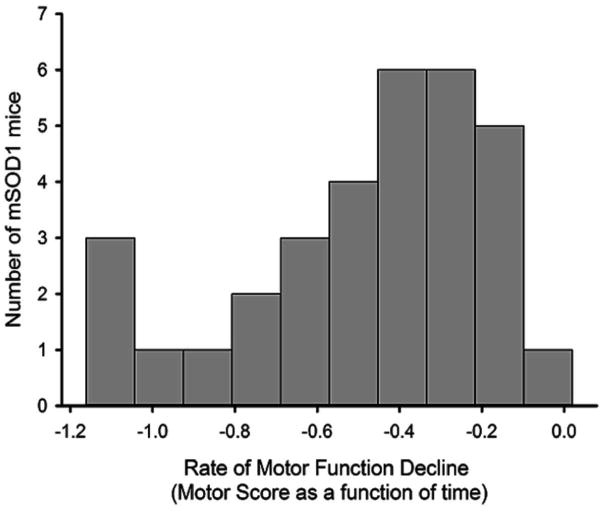
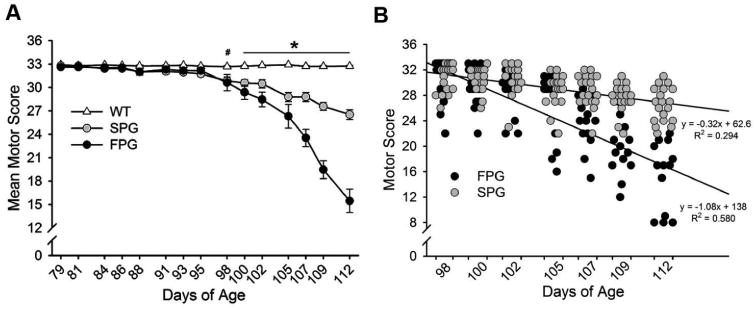
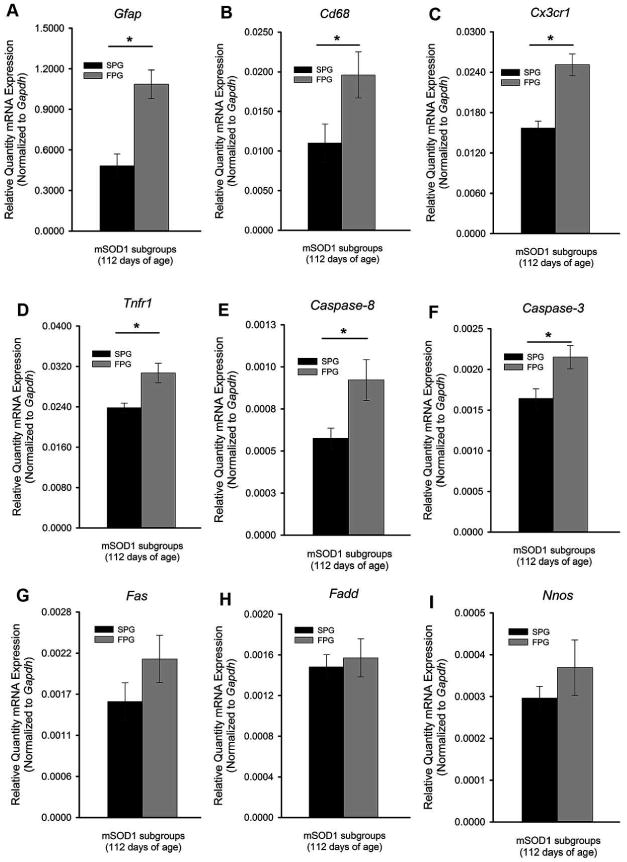
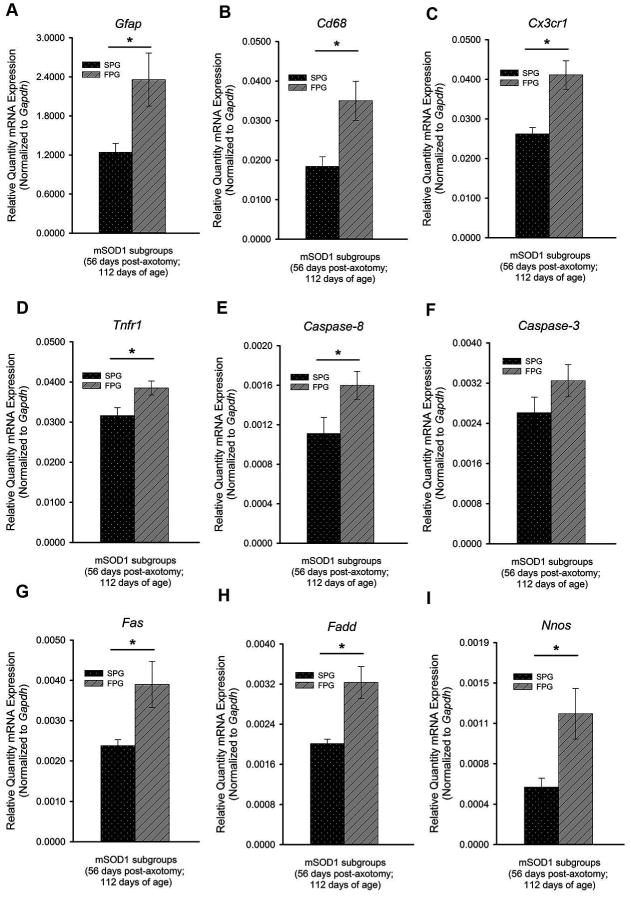
Disease progression rates among patients with amyotrophic lateral sclerosis (ALS) vary greatly. Although the majority of affected individuals survive 3-5 years following diagnosis, some subgroups experience a more rapidly progressing form, surviving less than 1 year, and other subgroups experience slowly progressing forms, surviving nearly 50 years. Genetic heterogeneity and environmental factors pose significant barriers in investigating patient progression rates. Similar to the case for humans, variation in survival within the mSOD1 mouse has been well documented, but different progression rates have not been investigated. The present study identifies two subgroups of B6SJL mSOD1(G93A) mice with different disease progression rates, a fast progression group (FPG) and slow progression group, as evidenced by differences in the rate of motor function decline. In addition, increased disease-associated gene expression within the FPG facial motor nucleus confirmed the presence of a more severe phenotype. We hypothesize that a more severe disease phenotype could be the result of 1) an earlier onset of axonal disconnection with a consistent degeneration rate or 2) a more severe or accelerated degenerative process. We performed a facial nerve transection axotomy in both mSOD1 subgroups prior to disease onset as a method to standardize the axonal disconnection. Instead of leading to comparable gene expression in both subgroups, this standardization did not eliminate the severe phenotype in the FPG facial nucleus, suggesting that the FPG phenotype is the result of a more severe or accelerated degenerative process. We theorize that these mSOD1 subgroups are representative of the rapid and slow disease phenotypes often experienced in ALS.
Keywords: ALS; MN; disease progression; facial nerve axotomy; gene expression; mSOD1; motoneuron.
© 2015 Wiley Periodicals, Inc.
Conflict of interest statement
Figures

References
-
- Abe K, Aoki M, Ikeda M, Watanabe M, Hirai S, Itoyama Y. Clinical characteristics of familial amyotrophic lateral sclerosis with Cu/Zn superoxide dismutase gene mutations. Journal of the neurological sciences. 1996;136(1-2):108–116. - PubMed
-
- Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain research Molecular brain research. 2004;130(1-2):7–15. - PubMed
-
- Armon C, Brandstater ME. Motor unit number estimate-based rates of progression of ALS predict patient survival. Muscle & nerve. 1999;22(11):1571–1575. - PubMed
-
- Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2000;1(5):293–299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
