

Molecular dynamics simulation of triclinic lysozyme in a crystal lattice
- PMID: 26013419
- PMCID: PMC4815314
- DOI: 10.1002/pro.2713
Molecular dynamics simulation of triclinic lysozyme in a crystal lattice
Abstract
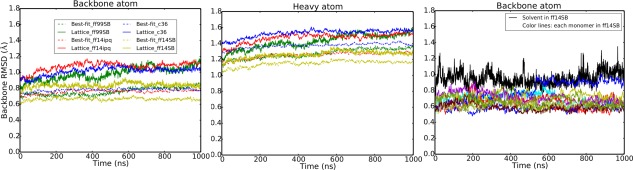
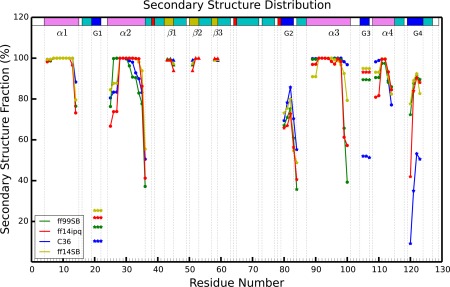
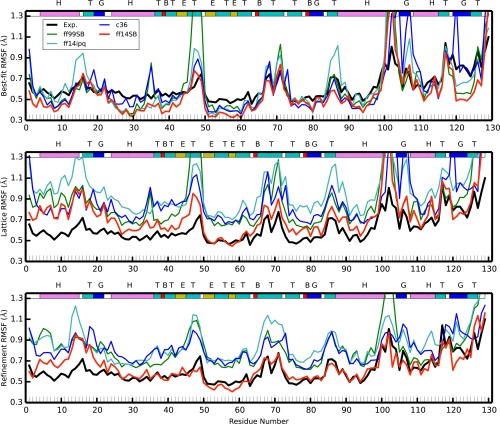
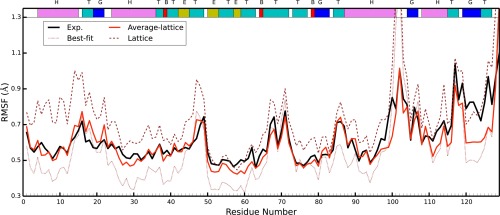
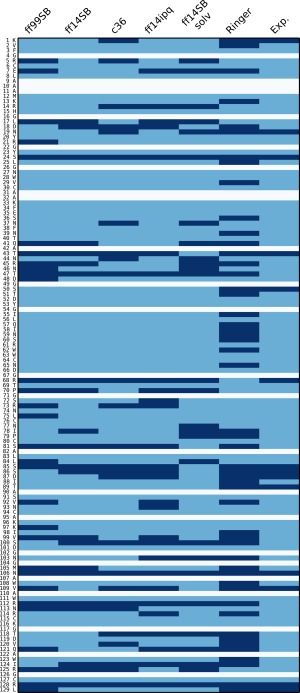
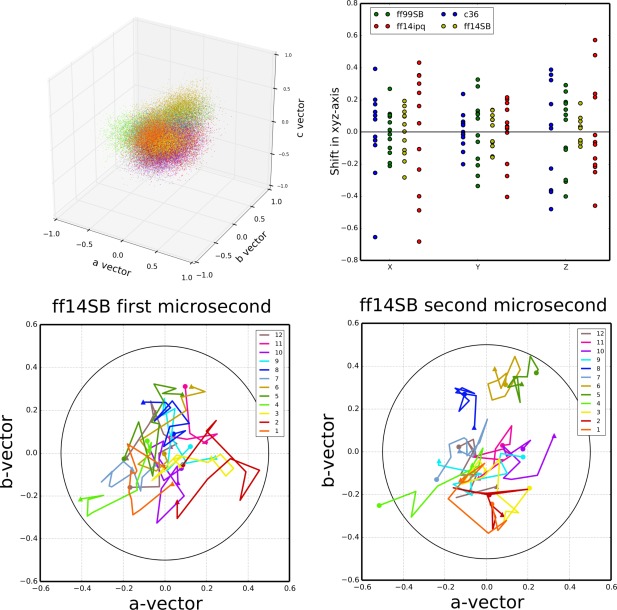
Molecular dynamics simulations of crystals can enlighten interpretation of experimental X-ray crystallography data and elucidate structural dynamics and heterogeneity in biomolecular crystals. Furthermore, because of the direct comparison against experimental data, they can inform assessment of molecular dynamics methods and force fields. We present microsecond scale results for triclinic hen egg-white lysozyme in a supercell consisting of 12 independent unit cells using four contemporary force fields (Amber ff99SB, ff14ipq, ff14SB, and CHARMM 36) in crystalline and solvated states (for ff14SB only). We find the crystal simulations consistent across multiple runs of the same force field and robust to various solvent equilibration schemes. However, convergence is slow compared with solvent simulations. All the tested force fields reproduce experimental structural and dynamic properties well, but Amber ff14SB maintains structure and reproduces fluctuations closest to the experimental model: its average backbone structure differs from the deposited structure by 0.37Å; by contrast, the average backbone structure in solution differs from the deposited by 0.65Å. All the simulations are affected by a small progressive deterioration of the crystal lattice, presumably due to imperfect modeling of hydrogen bonding and other crystal contact interactions; this artifact is smallest in ff14SB, with average lattice positions deviating by 0.20Å from ideal. Side-chain disorder is surprisingly low with fewer than 30% of the nonglycine or alanine residues exhibiting significantly populated alternate rotamers. Our results provide helpful insight into the methodology of biomolecular crystal simulations and indicate directions for future work to obtain more accurate energy models for molecular dynamics.
Keywords: biomolecular crystallography; computational crystallography; crystal simulations; force fields; lysozyme; molecular dynamics.
© 2015 The Protein Society.
Figures
References
-
- Henzler‐Wildman K, Kern D (2007) Dynamic personalities of proteins. Nature 450:964–972. - PubMed
-
- Kern D, Zuiderweg ER (2003) The role of dynamics in allosteric regulation. Curr Opin Struct Biol 13:748–757. - PubMed
-
- Schmidt A, Lamzin VS (2005) Extraction of functional motion in trypsin crystal structures. Acta Crystallogr D 61:1132–1139. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
