

Structure of a bacterial toxin-activating acyltransferase
- PMID: 26016525
- PMCID: PMC4466738
- DOI: 10.1073/pnas.1503832112
Structure of a bacterial toxin-activating acyltransferase
Abstract
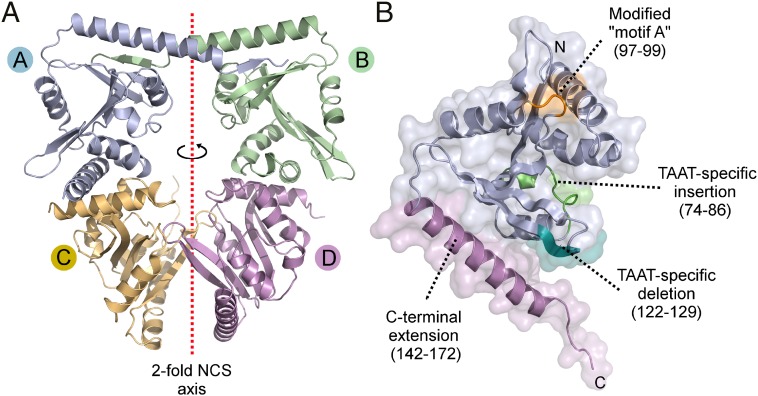
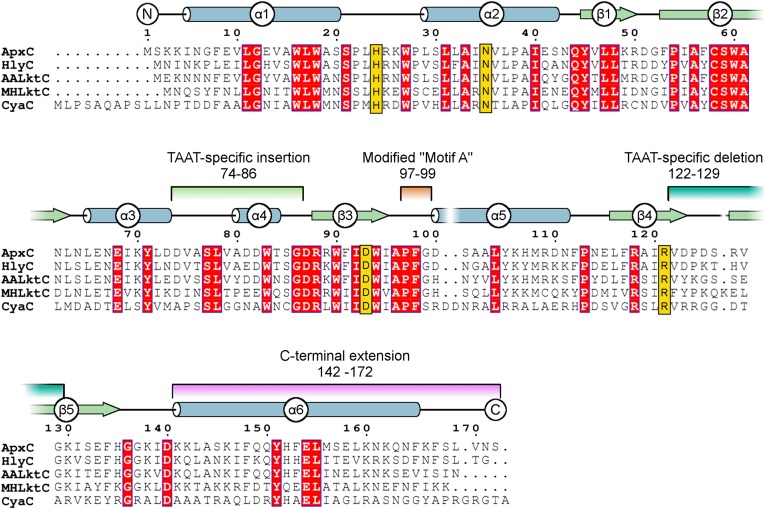
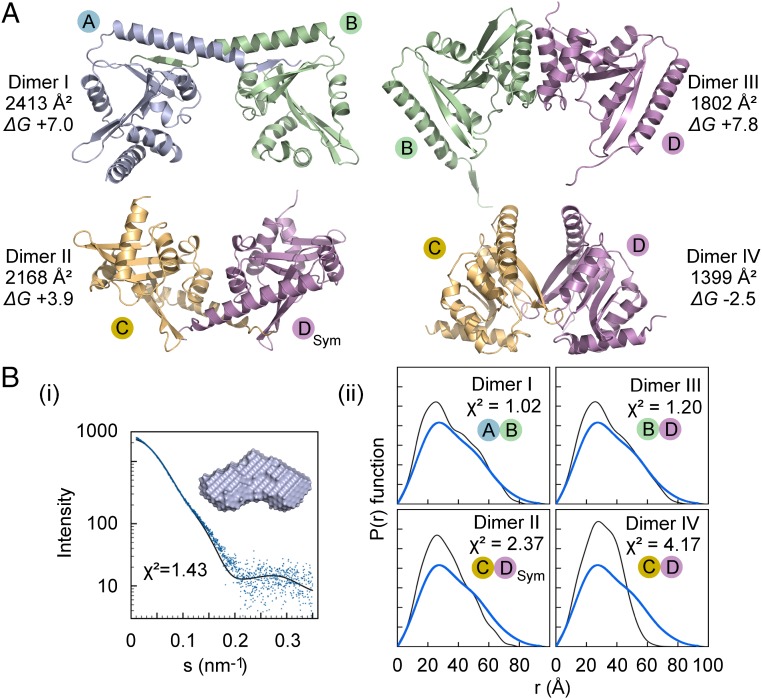
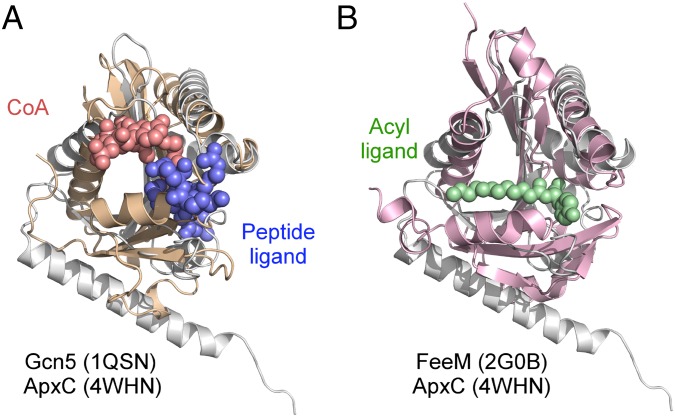
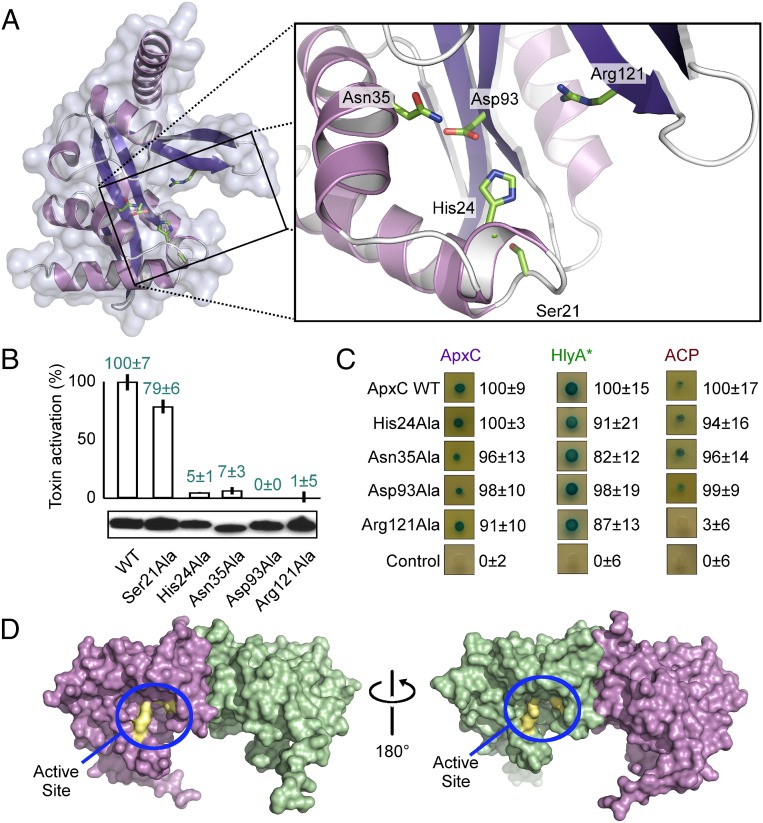
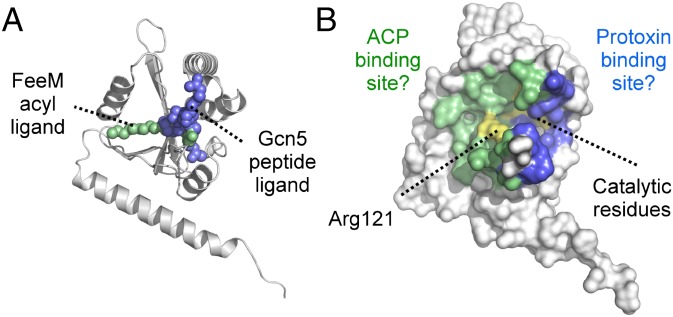
Secreted pore-forming toxins of pathogenic Gram-negative bacteria such as Escherichia coli hemolysin (HlyA) insert into host-cell membranes to subvert signal transduction and induce apoptosis and cell lysis. Unusually, these toxins are synthesized in an inactive form that requires posttranslational activation in the bacterial cytosol. We have previously shown that the activation mechanism is an acylation event directed by a specialized acyl-transferase that uses acyl carrier protein (ACP) to covalently link fatty acids, via an amide bond, to specific internal lysine residues of the protoxin. We now reveal the 2.15-Å resolution X-ray structure of the 172-aa ApxC, a toxin-activating acyl-transferase (TAAT) from pathogenic Actinobacillus pleuropneumoniae. This determination shows that bacterial TAATs are a structurally homologous family that, despite indiscernible sequence similarity, form a distinct branch of the Gcn5-like N-acetyl transferase (GNAT) superfamily of enzymes that typically use acyl-CoA to modify diverse bacterial, archaeal, and eukaryotic substrates. A combination of structural analysis, small angle X-ray scattering, mutagenesis, and cross-linking defined the solution state of TAATs, with intermonomer interactions mediated by an N-terminal α-helix. Superposition of ApxC with substrate-bound GNATs, and assay of toxin activation and binding of acyl-ACP and protoxin peptide substrates by mutated ApxC variants, indicates the enzyme active site to be a deep surface groove.
Keywords: X-ray crystallography; acyl carrier protein; acyltransferase; hemolysin; posttranslational modification.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Ulett GC, et al. Uropathogenic Escherichia coli virulence and innate immune responses during urinary tract infection. Curr Opin Microbiol. 2013;16(1):100–107. - PubMed
-
- Uhlén P, et al. Alpha-haemolysin of uropathogenic E. coli induces Ca2+ oscillations in renal epithelial cells. Nature. 2000;405(6787):694–697. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
