

Chromothripsis from DNA damage in micronuclei
- PMID: 26017310
- PMCID: PMC4742237
- DOI: 10.1038/nature14493
Chromothripsis from DNA damage in micronuclei
Abstract
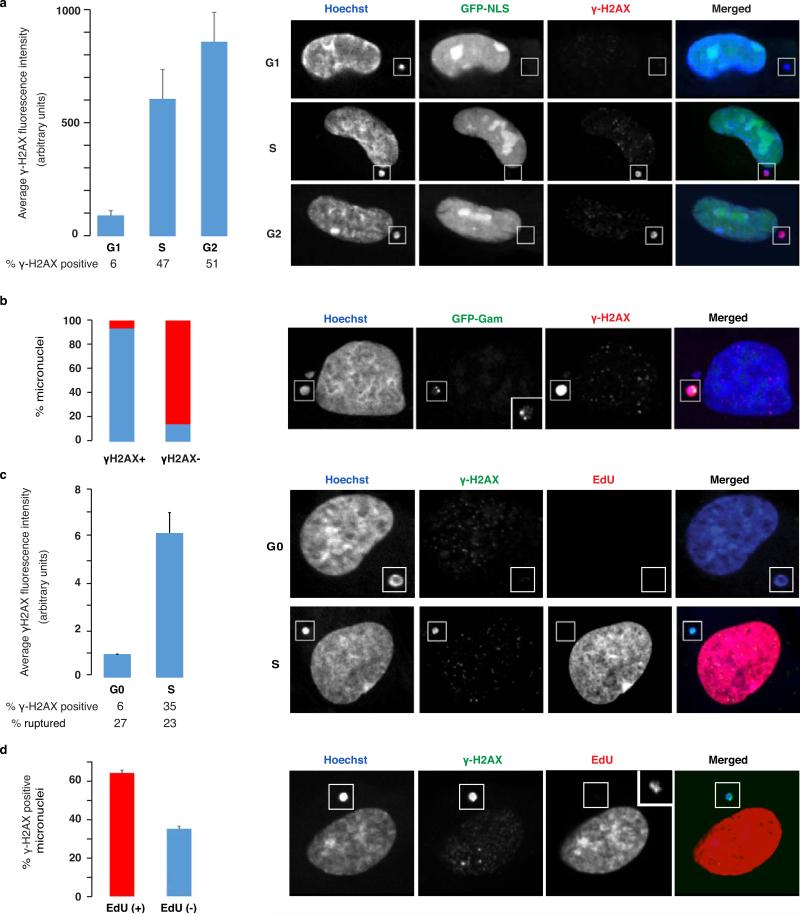
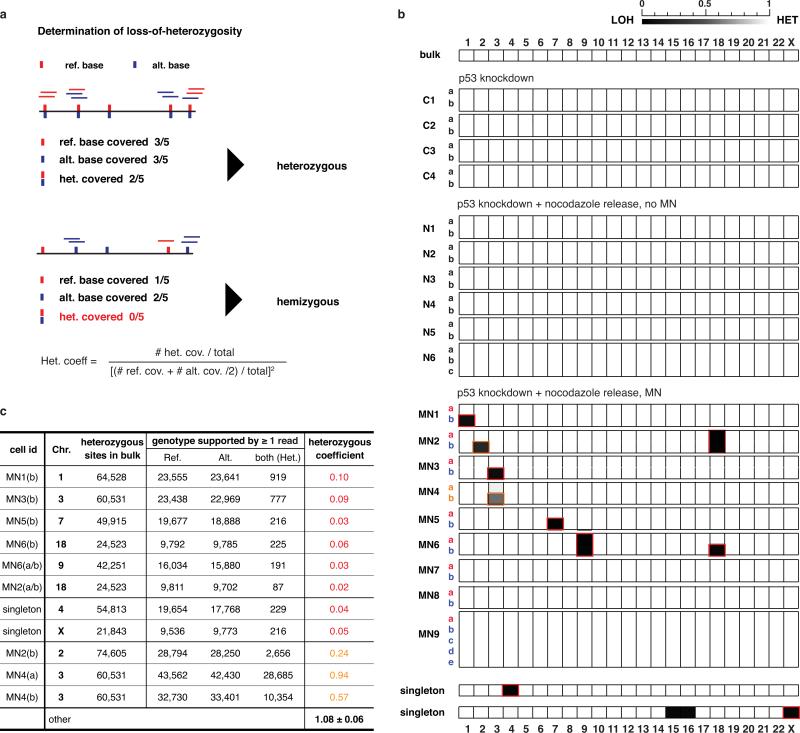
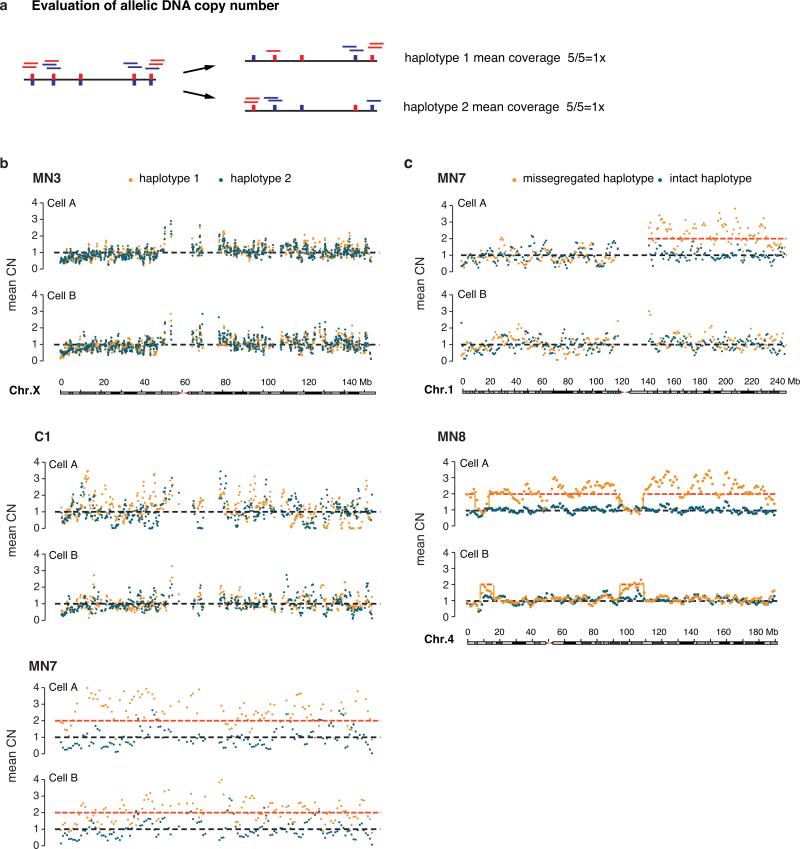
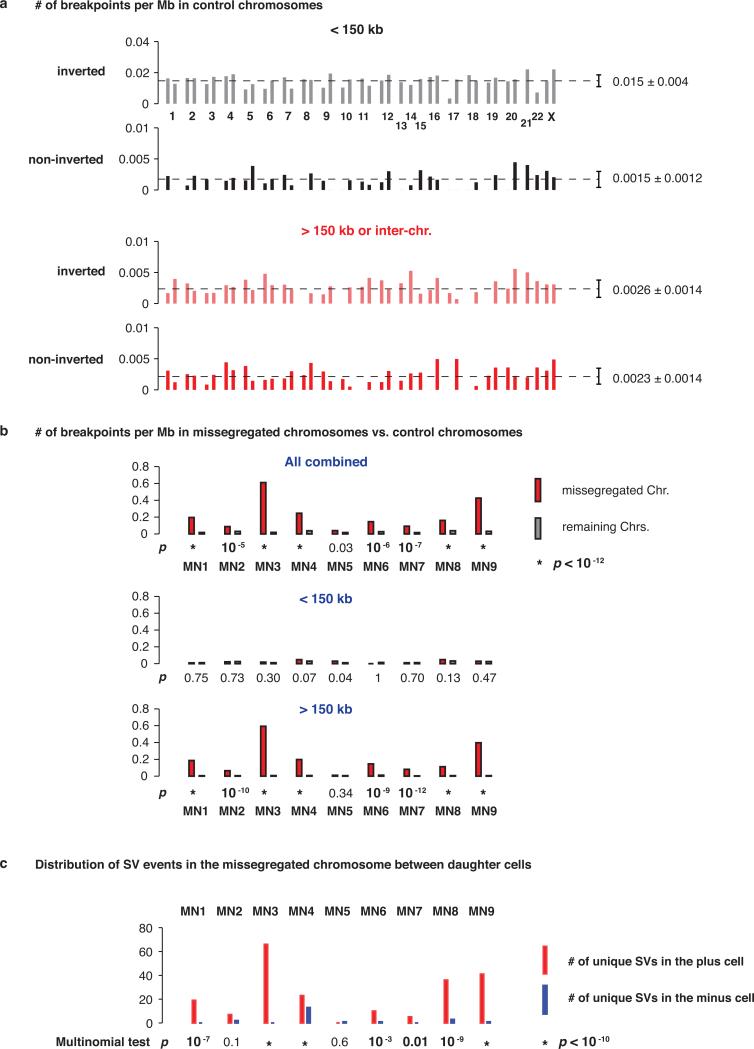
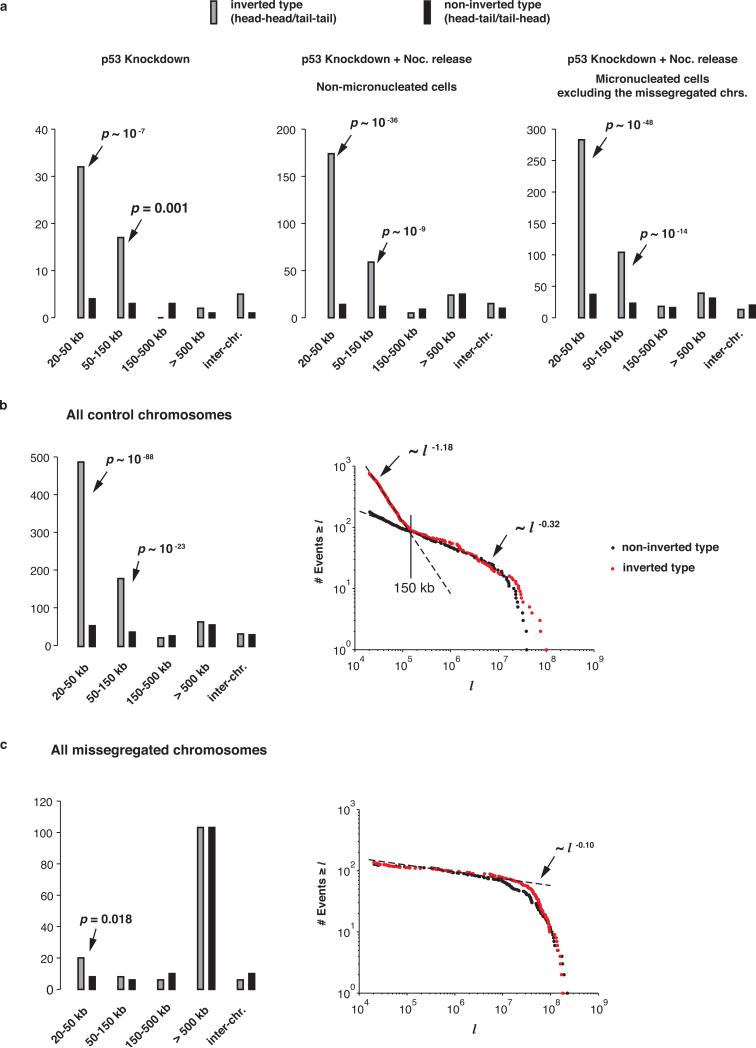
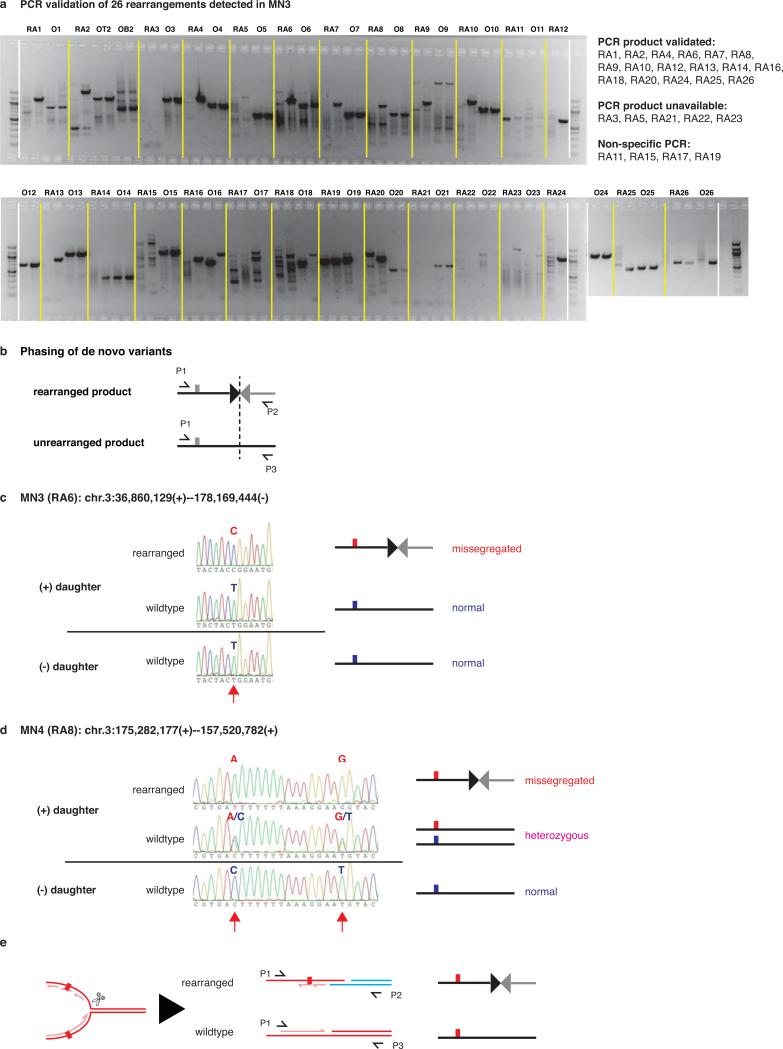
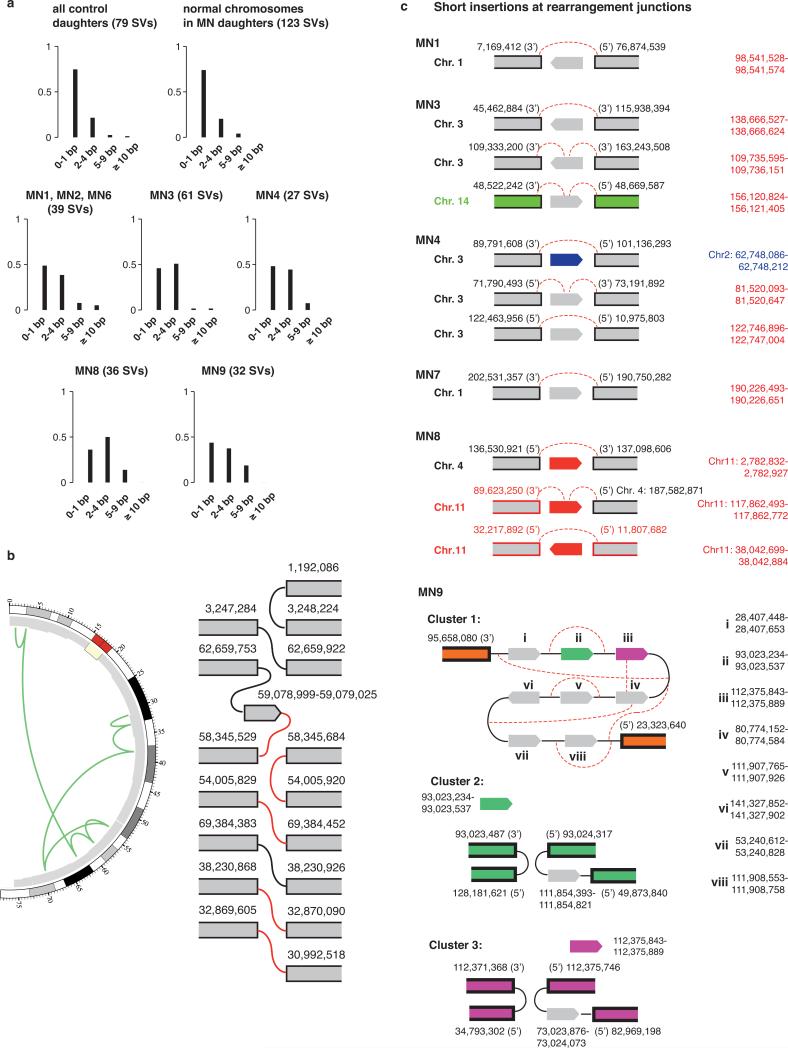
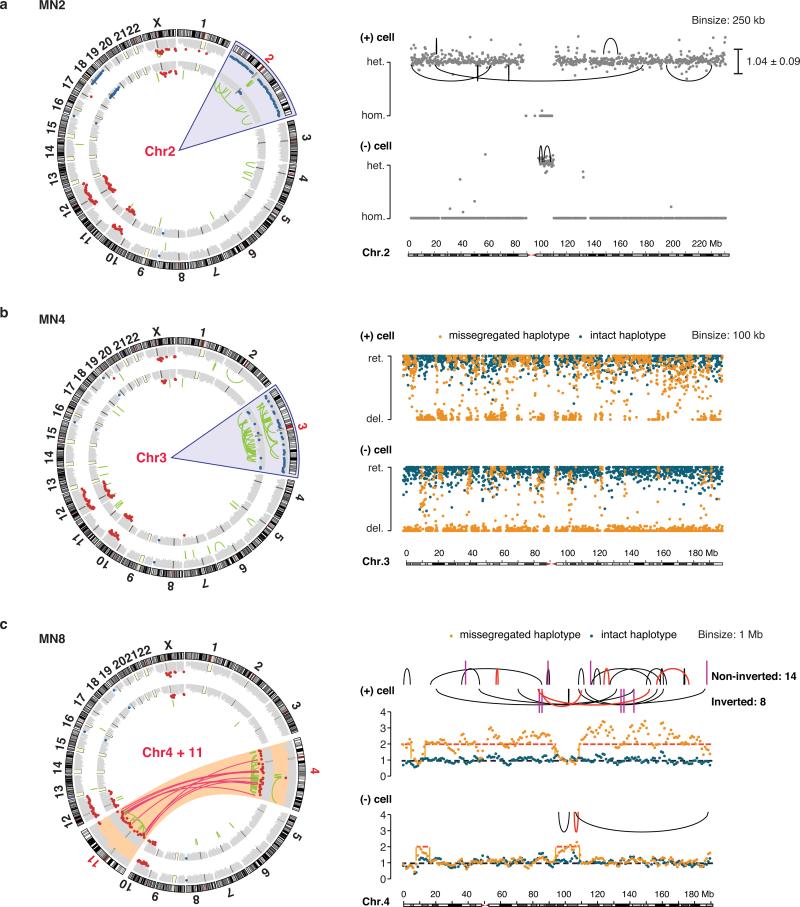
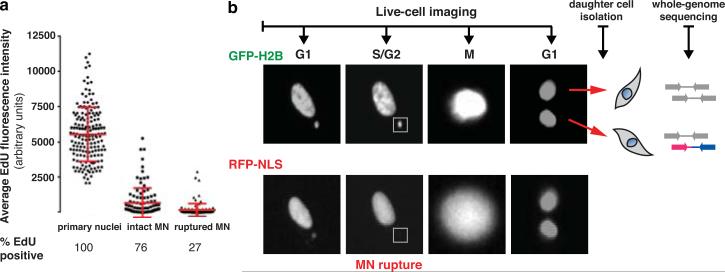
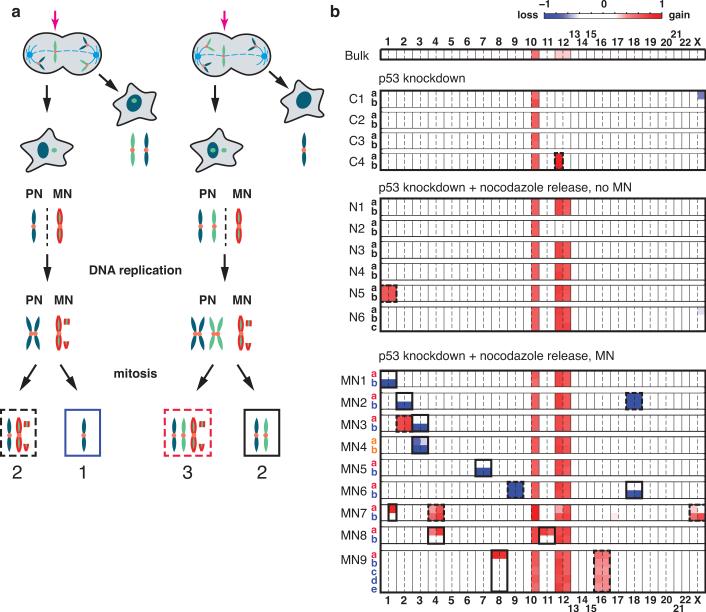
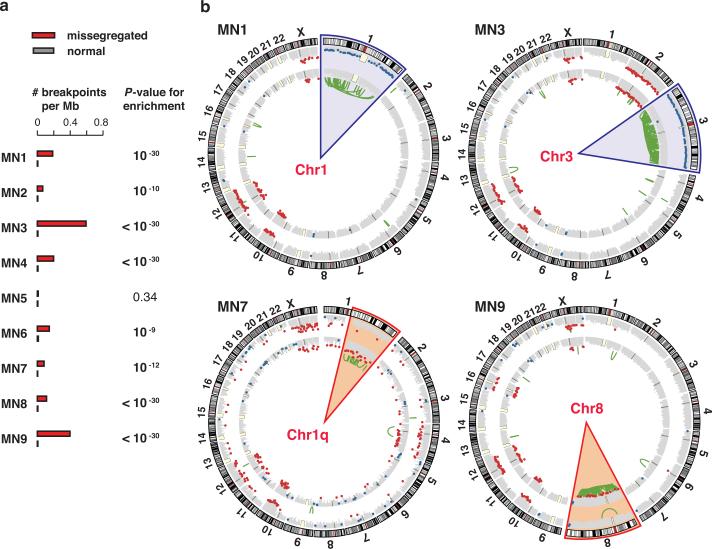
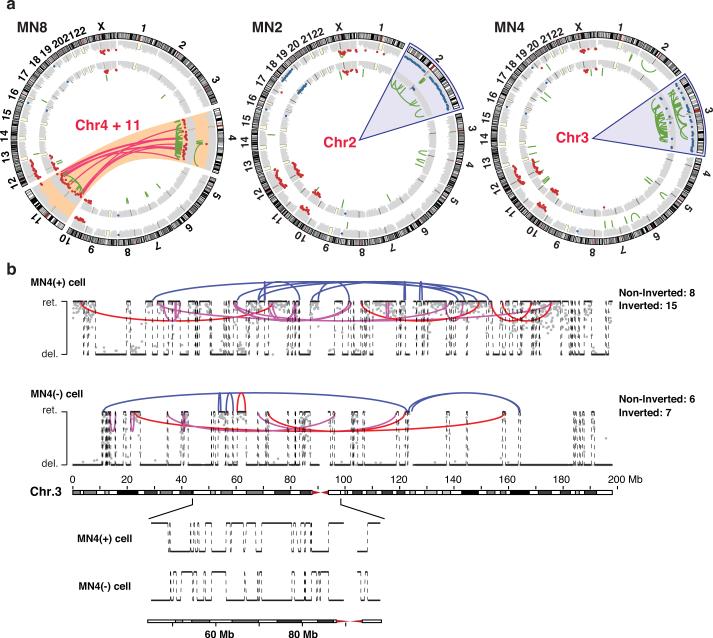
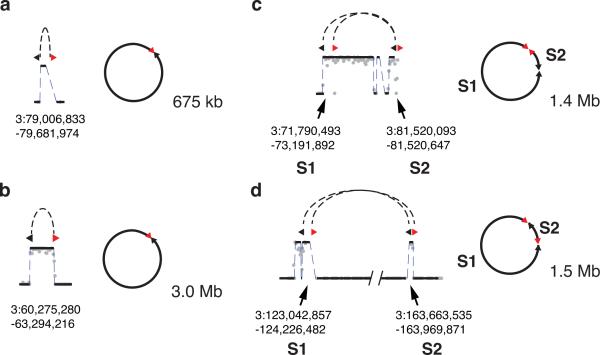
Genome sequencing has uncovered a new mutational phenomenon in cancer and congenital disorders called chromothripsis. Chromothripsis is characterized by extensive genomic rearrangements and an oscillating pattern of DNA copy number levels, all curiously restricted to one or a few chromosomes. The mechanism for chromothripsis is unknown, but we previously proposed that it could occur through the physical isolation of chromosomes in aberrant nuclear structures called micronuclei. Here, using a combination of live cell imaging and single-cell genome sequencing, we demonstrate that micronucleus formation can indeed generate a spectrum of genomic rearrangements, some of which recapitulate all known features of chromothripsis. These events are restricted to the mis-segregated chromosome and occur within one cell division. We demonstrate that the mechanism for chromothripsis can involve the fragmentation and subsequent reassembly of a single chromatid from a micronucleus. Collectively, these experiments establish a new mutational process of which chromothripsis is one extreme outcome.
Figures
Comment in
-
Cell biology: the micronucleus gets its big break.Nature. 2015 Jun 11;522(7555):162-3. doi: 10.1038/nature14528. Epub 2015 May 27. Nature. 2015. PMID: 26017308 No abstract available.
-
GENETICS. Making heads or tails of shattered chromosomes.Science. 2015 Jun 12;348(6240):1205-6. doi: 10.1126/science.aac5277. Science. 2015. PMID: 26068832 No abstract available.
-
Genome stability: Chromothripsis and micronucleus formation.Nat Rev Genet. 2015 Jul;16(7):376-7. doi: 10.1038/nrg3970. Nat Rev Genet. 2015. PMID: 26084493 No abstract available.
-
Linking Micronuclei to Chromosome Fragmentation.Cell. 2015 Jun 18;161(7):1502-4. doi: 10.1016/j.cell.2015.06.005. Cell. 2015. PMID: 26091034
-
Understanding Complexity of Cancer Genomes: Lessons from Errors.Dev Cell. 2020 Jun 8;53(5):500-502. doi: 10.1016/j.devcel.2020.05.004. Dev Cell. 2020. PMID: 32516595
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
