

Evidence Suggesting That Discontinuous Dosing of ALK Kinase Inhibitors May Prolong Control of ALK+ Tumors
- PMID: 26018086
- PMCID: PMC4506255
- DOI: 10.1158/0008-5472.CAN-14-3437
Evidence Suggesting That Discontinuous Dosing of ALK Kinase Inhibitors May Prolong Control of ALK+ Tumors
Abstract
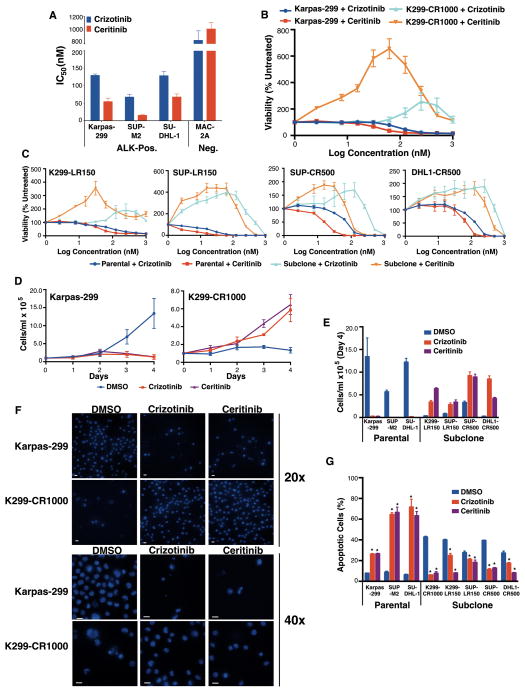
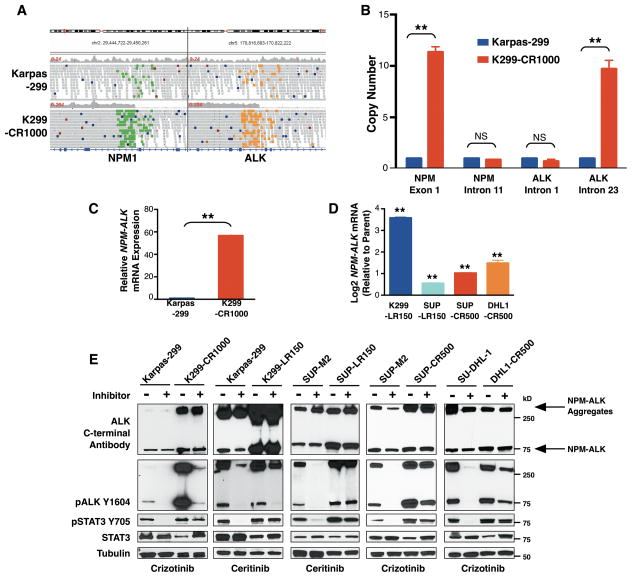
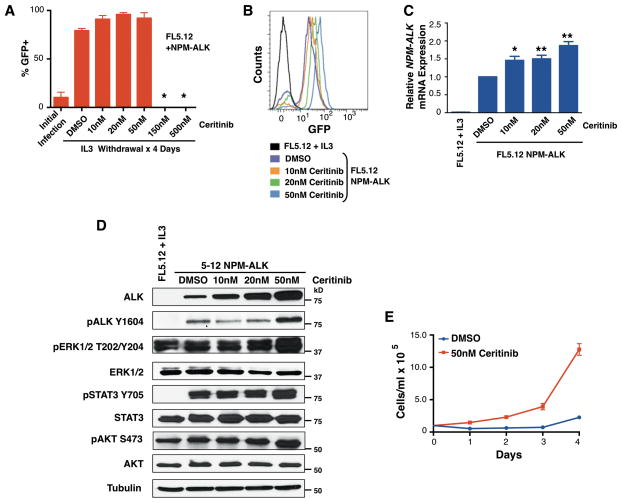
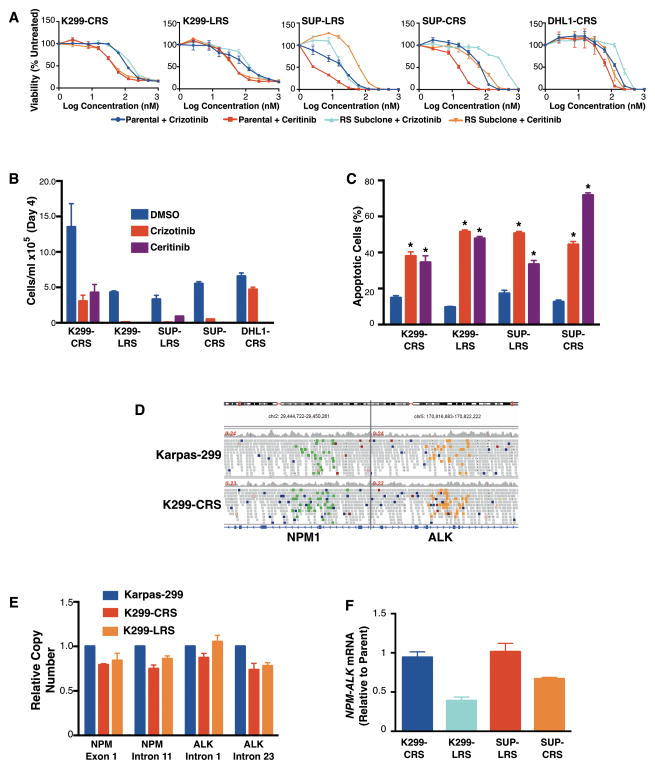
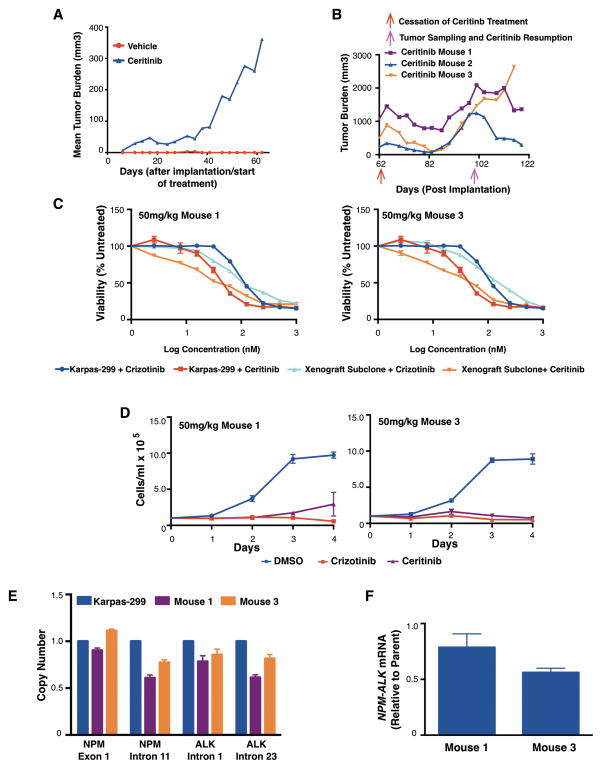
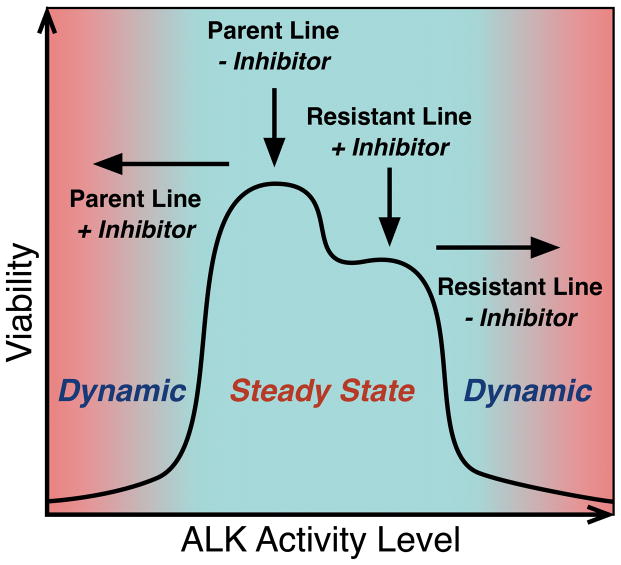
The anaplastic lymphoma kinase (ALK) is chromosomally rearranged in a subset of certain cancers, including 2% to 7% of non-small cell lung cancers (NSCLC) and ∼70% of anaplastic large cell lymphomas (ALCL). The ALK kinase inhibitors crizotinib and ceritinib are approved for relapsed ALK(+) NSCLC, but acquired resistance to these drugs limits median progression-free survival on average to ∼10 months. Kinase domain mutations are detectable in 25% to 37% of resistant NSCLC samples, with activation of bypass signaling pathways detected frequently with or without concurrent ALK mutations. Here we report that, in contrast to NSCLC cells, drug-resistant ALCL cells show no evidence of bypassing ALK by activating alternate signaling pathways. Instead, drug resistance selected in this setting reflects upregulation of ALK itself. Notably, in the absence of crizotinib or ceritinib, we found that increased ALK signaling rapidly arrested or killed cells, allowing a prolonged control of drug-resistant tumors in vivo with the administration of discontinuous rather than continuous regimens of drug dosing. Furthermore, even when drug resistance mutations were detected in the kinase domain, overexpression of the mutant ALK was toxic to tumor cells. We confirmed these findings derived from human ALCL cells in murine pro-B cells that were transformed to cytokine independence by ectopic expression of an activated NPM-ALK fusion oncoprotein. In summary, our results show how ALK activation functions as a double-edged sword for tumor cell viability, with potential therapeutic implications.
©2015 American Association for Cancer Research.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–49. - PubMed
-
- Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene expression patterns : GEP. 2006;6:448–61. - PubMed
-
- Crockett DK, Lin Z, Elenitoba-Johnson KS, Lim MS. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene. 2004;23:2617–29. - PubMed
-
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nature reviews Cancer. 2013;13:685–700. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
