

doi: 10.1186/s13073-015-0167-x.
eCollection 2015.
JAFFA: High sensitivity transcriptome-focused fusion gene detection
Affiliations
- PMID: 26019724
- PMCID: PMC4445815
- DOI: 10.1186/s13073-015-0167-x
Item in Clipboard
JAFFA: High sensitivity transcriptome-focused fusion gene detection
Genome Med.
.
Abstract
Genomic instability is a hallmark of cancer and, as such, structural alterations and fusion genes are common events in the cancer landscape. RNA sequencing (RNA-Seq) is a powerful method for profiling cancers, but current methods for identifying fusion genes are optimised for short reads. JAFFA (https://github.com/Oshlack/JAFFA/wiki) is a sensitive fusion detection method that outperforms other methods with reads of 100 bp or greater. JAFFA compares a cancer transcriptome to the reference transcriptome, rather than the genome, where the cancer transcriptome is inferred using long reads directly or by de novo assembling short reads.
Figures
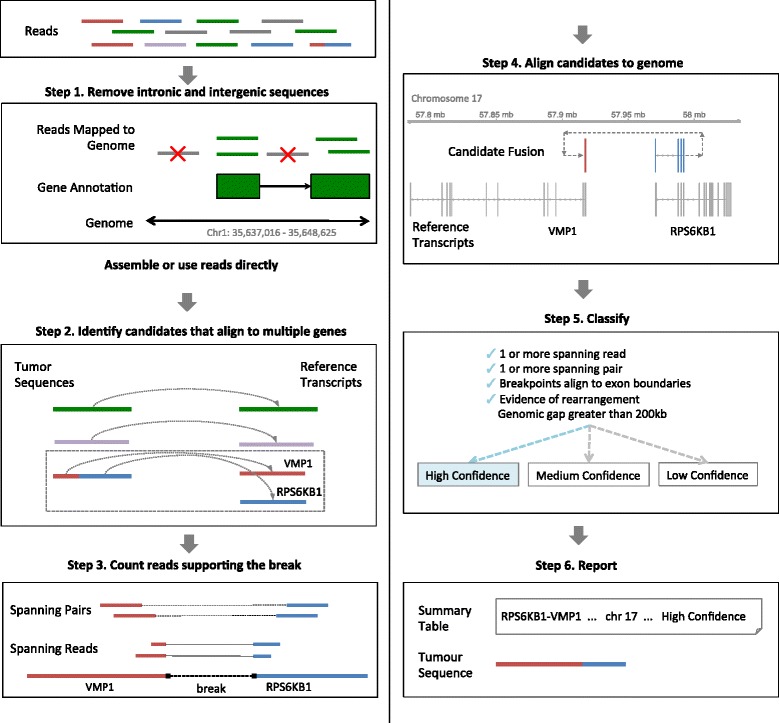
The JAFFA pipeline. An example of the JAFFA pipeline is demonstrated in detail using the RPS6KB1-VMP1 fusion from the MCF-7 breast cancer cell line dataset. Step 1: RNA-Seq reads are first filtered to remove intronic and intergenic reads. 50 bp reads would then be assembled into contigs using Oases. For longer reads, this step is not necessary. Step 2: The resulting tumour sequences are then aligned to the reference transcriptome and those that align to multiple genes are selected. These contigs make up a set of initial candidate fusions. Step 3: Next, the pipeline counts the number of reads and read pairs that span the breakpoint. Step 4: Candidates are then aligned to the human genome. Genomic coordinates of the breakpoint are determined. Step 5: Further selection and candidate classification is carried out using quantities such as genomic gap size, supporting reads and alignment of breakpoints to exon-exon boundaries. Step 6: A final list of candidates is reported along with their sequence.
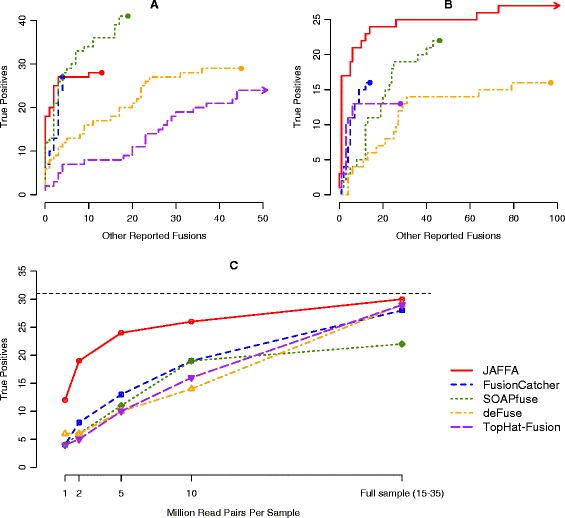
Performance of JAFFA and four other tools on cancer RNA-Seq. (A) A ROC-style curve for the ranking of candidate fusions in the Edgren dataset. The Edgren dataset consists of between 7 and 21 million 50 bp read pairs of the BT-474, SK-BR-3, KPL-4 and MCF-7 cell lines. The number of true positives fusions are plotted against the number of other reported fusions from a ranked list of fusion candidates. Probable true positives (see text for detail) are removed. Higher curves indicate a better ranking of the true positives. For each fusion detection tool, we ranked the candidates using the tools own scoring system, or if absent, the supporting data that maximised the area under the curve. SOAPfuse ranked true positives higher than other tools, followed by FusionCatcher and JAFFA. (B) On long read data - the ENCODE dataset consisting of 20 million 100 bp read pairs of the MCF-7 cell line - JAFFA ranks true positives higher than any other tool. (C) JAFFA’s sensitivity is confirmed on a second long read dataset - 13 glioma samples with read depths in the range of 15 to 35 million 100 bp read-pairs. JAFFA identifies 30 of the 31 true positives (total true positives are indicated by the dashed line). Downsampling the data to mimic smaller read depths indicates that JAFFA has similar sensitivity with 2 million read pairs per samples as other tools on 10 million read pairs per sample.
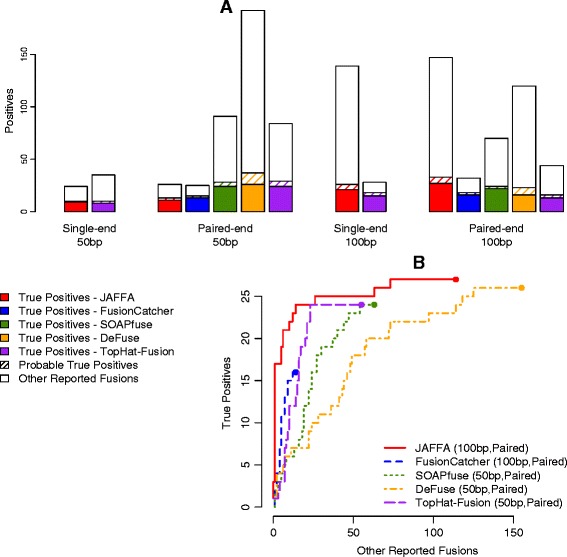
Performance of JAFFA and four other tools for different read lengths and layouts. We compared the performance of JAFFA, FusionCatcher, SOAPfuse, deFuse and TopHat-Fusion on the ENCODE dataset of the MCF-7 cell line, trimmed to emulate four different read configurations: single-end 50 bp (80 million reads), paired-end 50 bp (40 million read pairs), single-end 100 bp (40 million reads) and paired-end 100 bp (20 million read pairs). In each case, the total number of bases sequence was 4 billion. Only JAFFA and TopHat-Fusion could process single-end data. (A) Most true positives were reported with JAFFA on 100 bp paired-end reads followed by deFuse on 50 bp paired-end reads. (B) For each tool we compared the ranking of fusions, by selecting the read length and layout that maximised ROC performance. We ranked the candidates using the tools own scoring system, or if absent, the supporting data that maximised the area under the curve. JAFFA on 100 bp reads ranked true positives higher than any other combination.
References
-
- Edwards PAW. Fusion genes and chromosome translocations in the common epithelial cancers. J Pathol. 2010;220:244–54. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
